Oncomedicine 2017; 2:156-167. doi:10.7150/oncm.22477 This volume Cite
Review
Stressed Out - Therapeutic Implications of ER Stress Related Cancer Research
Randal Riha1, Pooja Gupta-Saraf1, Payel Bhanja1, Samyak Badkul1, Subhrajit Saha1, 2 ![]()
1. Department of Radiation Oncology, University of Kansas Medical Center, Kansas 66160, USA;
2. Department of Cancer Biology, University of Kansas Medical Center, Kansas 66160, USA.
Received 2017-8-22; Accepted 2017-9-25; Published 2017-9-27
Abstract

The unfolded protein response (UPR) is an established and well-studied cellular response to the stress and serves to relieve the stress and reinstate cellular homeostasis. It occurs in the endoplasmic reticulum (ER), responsible of properly folding and processing of secretory and transmembrane proteins. It is extremely sensitive to alteration in homeostasis caused by various internal or external stressors which leads to accumulation of misfolded or unfolded proteins in the ER lumen. The UPR works by restoring protein homeostasis in the ER, either through the boosting of protein-folding and degradation capability or by assuaging the demands for such effects, and can cause the activation of cell death if unable to do so. Cancer cells have adapted to gain advantage from the UPR and keeping the cell away from apoptosis and promoting survival, including survival of the cancer stem cells and evading the immune system. Several components of the UPR are overexpressed in a malignant cell and are responsible for resistance from various chemotherapy options and radiotherapy, which are also responsible for causing ER stress and activating the UPR. In this review, we discuss the various ways in which UPR can aid different cancers to survive and evade therapy and highlight recent research, which exploits the UPR to confer sensitivity to these cancer cells against various drugs and radiation.
Keywords: ER Stress, UPR, Cancer Stem Cells, Radiation, Immunotherapy.
Introduction
The ability to survive and proliferate in a hostile biological environment is a hallmark of cancer. In the context of rapidly proliferating cells, there is a large demand for protein synthesis. The endoplasmic reticulum (ER) is the key organelle for proper synthesis and folding of these proteins. Many insults are capable of disturbing ER homeostasis, resulting in diminished capacity for proper protein folding, including hypoxia, nutrient starvation, acidosis, redox imbalance, loss of calcium homeostasis, or exposure to drugs or other compounds. The result is a buildup of unfolded and improperly folded proteins, termed ER stress. Cells react to the buildup of these proteins by activating defense pathways termed the unfolded protein response (UPR), which function to reestablish ER homeostasis, aid in adaptation, as well as initiating apoptosis in the scenario of excessive ER stress. Malignant cells rely on these UPR pathways to adapt to perturbations in ER folding capacity due to the hostile tumor microenvironment as well as the increase in unfolded and misfolded proteins resulting from high synthetic demands. Through study of these pathways in cancer, it is becoming increasingly clear that the UPR can be therapeutically exploited to reduce the survivability of malignant cells, or tip the balance towards apoptosis. In this review, we have discussed studies on UPR signaling in cancer research and their applications in cancer therapy.
ER Stress and the Unfolded Protein Response
There are three ER transmembrane receptors which govern the UPR. They are pancreatic ER kinase (PERK), activating transcription factor 6 (ATF6), and inositol requiring enzyme 1 (IRE1). These three receptors are maintained in an inactive state during ER homeostasis by interaction with glucose-regulated protein 78 kDa (GRP78), which is also known as binding immunoglobulin protein (BiP) and heat shock 70 kDa protein 5 (HSPA5). GRP78 is a molecular chaperone which functions to bind to and promote folding of unfolded proteins in the ER lumen. These unfolded proteins are thought to compete for GRP78 binding with the ER luminal domains of the stress receptors. GRP78 dissociation from the ER transmembrane receptors subsequently activates or positively regulates components of the UPR [1, 2].
PERK activation results in phosphorylation of eukaryotic translation initiation factor alpha subunit (eIF2α), which inhibits protein translation, reducing the ER stress burden [3, 4]. Cap independent mRNAs such as activating transcription factor 4 (ATF4) are preferentially translated leading to transcriptional activation of GRP78, growth arrest and DNA damage-inducible protein transcript 34 (GADD34), and the pro-apoptotic protein C/EBP homologous protein (CHOP) [5-10]. While initially contributing to cellular survival in conditions of ER stress, PERK is considered pro-apoptotic due to strong induction of CHOP in chronic or terminal ER stress. ATF6 dissociates from GRP78 during periods of ER stress, after which, it translocates to the Golgi apparatus where it is cleaved by protease, allowing translocation to the nucleus [2, 11]. ATF6 acts on the promoter region of UPR target genes termed the endoplasmic reticulum stress element (ERSE), activating genes responsible for components of the UPR response, including molecular chaperones GRP78, Grp94, and calreticulin, as well as CHOP, and XBP1 [12-15]. ATF6 is primarily considered pro-survival due to its role in promoting transcription of chaperones and XBP1. IRE1, upon activation, splices XBP1 into the active form, producing a potent transcription factor that translocates to the nucleus, and binds to the ERSE to upregulate transcription of UPR genes, similarly to ATF6. One unique gene activated by XBP1 is that of ER degradation enhancing α-mannosidase-like protein (EDEM), which locates to the ER and functions as part of the ER associated degradation (ERAD) pathway, targeting misfolded proteins for export into the cytoplasm for degradation via the ubiquitin-proteasome pathway [16-19]. IRE1 is considered pro-survival, but can contribute to apoptotic signaling through the TRAF2-ASK-JNK pathway [20-22]. The actions of each branch of the UPR can be thought to contribute to a balance between survival and cell death (Figure 1).
Stem Cells and ER Stress
Stem cells are critical for maintenance of tissues and cell populations throughout the body. In addition to normal human stem cells, cancer stem cells (CSCs) are a population of cells that possess characteristics of normal stem cells, as well as capacity for tumorigenesis which make these cells capable of causing cancer recurrence and metastasis. Though there is substantial controversy regarding CSCs, it is clear, that they play a role in the heterogeneity of cells which make up a cancer. In addition to illuminating a role in the development, survival, recurrence, and metastasis of cancer cells, an understanding of how ER stress effects stem cells may uncover a target against these CSCs. The effect of ER stress on normal stem cell populations must also be considered in the setting of systemic or regional therapy utilizing ER stress inducing agents as there may be a potential for side effects.
CSCs have been demonstrated to be particularly resistant to traditional cancer therapy. The ability of these cells to seed tumors has led some to theorize that these cells are the origin of cancer relapse and metastasis [23]. Evidence is starting to suggest that the UPR protein GRP78 may have a role in the function and survival of these cells, which is not unexpected, as GRP78 is required for embryonic stem cell survival and maintenance of hematopoietic stem cells [24, 25]. One study showed MDA-468 breast cancer cells with a stem cell/progenitor phenotype having increased expression of UPR proteins GRP78, GRP94, and protein disulfide-isomerase. The presence of these proteins was confirmed to be present in breast cancer patient bone marrow when compared to disease-free control subjects [26]. Another study found GRP78 to play a crucial role in stem cell oncogenesis in MCF-7 breast cancer cells [27]. GRP78 has also been shown to play a critical role in tumorigenesis of prostate adenocarcinoma in mice with PTEN tumor suppressor gene deletion [28]. In human prostate cancer patients, GRP78 is upregulated and is correlated with clinical tumor recurrence and reduced survival [29]. Increased recurrence and reduced survival would be consistent with the presence of a population of treatment resistant CSCs. With the hypoxic niche of the bone marrow as a common site of metastatic cancer growth in breast and prostate cancer, it is intriguing to consider whether increased expression of GRP78 is a result of hypoxia related ER stress induction of the UPR, or whether GRP78 is induced by other processes, predisposing those cells to survival in that microenvironment. Whether hypoxia drives the processes that establish CSC phenotype is certainly an open question [30].
With the expression of GRP78 in CSCs, it may be a future target for eliminating stem cell populations responsible for recurrence and metastasis. In head and neck cancer initiating cells (HN-CICs), GRP78 has been found to contribute to maintenance of the cancer initiating cell population. By knocking down GRP78, tumor initiating activity was found to be lessened, however, not entirely eliminated [31]. More recently, two unique treatments have been shown promise through targeting GRP78. A monoclonal antibody against GRP78 was shown to suppress signaling through the PI3K/Akt/mTOR pathway, which is responsible for radiation resistance in non-small cell lung cancer and glioblastoma multiforme (GBM). They found that ionizing radiation increased GRP78 expression through the induction of ER stress, and treatment with the monoclonal antibody along with ionizing radiation in mouse xenograph models showed significant tumor growth delay [32]. The other recent therapeutic model utilized a phage, displaying a ligand specific to GRP78, to deliver the herpes simplex virus thymidine kinase type-1 suicide inducing transgene to mice with MDA-PCa-118b patient derived tumor xenografts. After treatment with the antiviral drug ganciclovir, tumors were reduced by an average of 50% [33]. It is curious to speculate whether ionizing radiation could be employed prior to treatment with the viral phage to upregulate expression of GRP78 to increase the effectiveness of phage targeting of cancer cells.
The effect of ER stress on normal stem cells must be considered in the context of ER stress related therapies. Hematopoietic stem cells have been shown to be predisposed to apoptosis through a strong PERK mediated response to ER stress. This is hypothesized to be a quality control mechanism to ensure clonal integrity of hematopoietic stem cells [34]. Although this indicates that caution should be warranted to avoid loss of this stem cell population, it also provides hope of potential therapy for hematopoietic malignancies.
Balance between survival and death. ER stress and the UPR can be considered as a balance between survival and cell death. A cellular insult inducing ER stress is countered by the UPR, resulting in a number of outcomes that can be simplified to three outcomes. A strong UPR, or minor insult results in adaptation, which is often used by cancer cells for survival. With increasing insult, a chronic ER stress can result in a balance that can be tipped towards apoptosis through further induction of ER stress. Finally, a terminal ER stress response ends in apoptotic cell death.
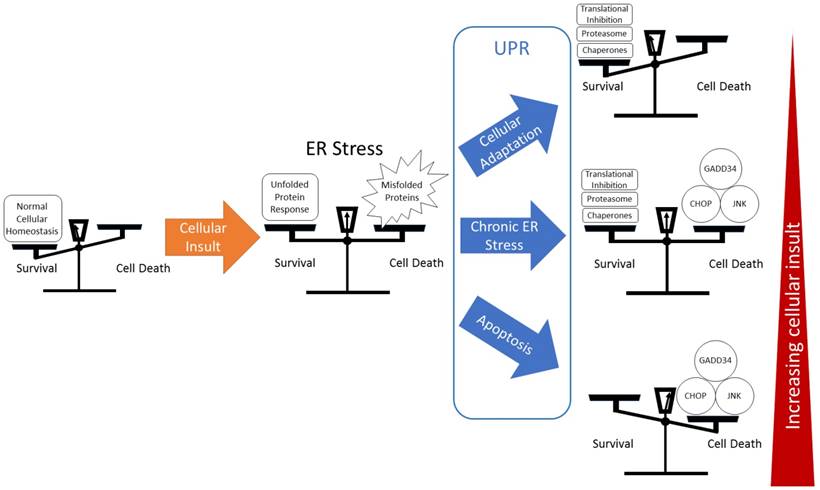
In the development of tumors and corresponding CSCs, mutations arise in a way that is unique to that specific cancer. The role that ER stress plays in the tumorigenesis is expected to vary between different types of cancer. Similarly, potential therapeutic targeting of ER stress and UPR pathways would be expected to differ based upon the underlying biology of each individual cancer.
Cancer therapy and ER stress
Recent cancer research has illuminated the many roles that the UPR play in the survival and propagation of cancer cells. As the UPR is being linked to survival adaptation in many diverse types of cancer cells, we are learning that there is a similarly diverse variability in activation or upregulation of the three major UPR pathways. As activation and reliance upon the UPR for survival differs by cell type and environmental niche, research into therapeutics to target the UPR are likely to be with specificity to cell type rather than a universal “magic bullet”. The variability of ER stress status and reliance on the UPR can also be considered as a potential biomarker useful for determining the most effective treatments and therapeutics. If a cancer is known to have a significant chronic ER stress, the clinician could choose a therapeutic most likely to result in a terminal ER stress response.
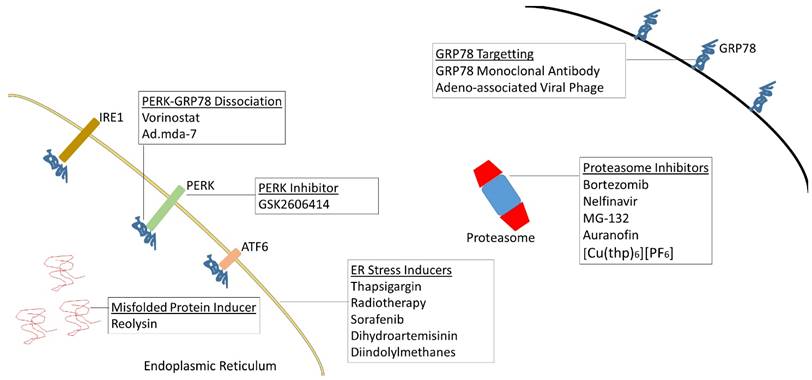
Table 1 illustrates the wide variety of cancer types that have been investigated in the context of the UPR. Figure 2 illustrates potential therapeutics which either induce ER stress or otherwise target the UPR. The following sections will highlight the aspects of ER stress and therapeutic implications unique to specific types of malignancies.
Hodgkin cells
Cells from Hodgkin's disease have been shown to have a defect in regulation of NF-κB. When treated with proteasome inhibitor MG-132, HD-My-Z Hodgkin cells showed no decrease in expression of NF-κB, likely due to constitutive activity in this cell line. These treated cells, however, underwent apoptosis, and radiosensitivity was noted when compared to intrinsic radiosensitivity in clonogenic survival assays. This radiosensitivity result is intriguing as NF-κB has been implicated in protection from apoptosis following exposure to ionizing radiation [35]. A look into the more recent research, may suggest ER stress and downstream UPR pathways as causal pathways for this increased radiosensitivity.
Cancers with ER stress related research
| Type of Cancer | Research demonstrating UPR for survival or carcinogenesis (references) | Research regarding ER stress in treatment (references) |
|---|---|---|
| Hodgkin's lymphoma | [36] | [37] |
| Pancreatic Adenocarcinoma | [39] | [41-44] |
| Hepatocellular Carcinoma | [48, 55] | [57, 60] |
| Prostate Adenocarcinoma | [29, 102-104] | [105, 106] |
| Colorectal Adenocarcinoma | [107] | [108-111] |
| Non-small Cell Lung Cancer | [112, 113] | [114, 115] |
| Ductal Carcinoma | [116, 117] | [118-120] |
Targets for therapy. ER stress and the UPR can be targeted at multiple sites. The potential implication is effective therapeutics for various types of malignancies
Hodgkin cells have been shown to have upregulated levels of UPR signaling molecules, specifically those that are associated with survival, including ATF6, GRP78, and XBP1 (both spliced and un-spliced). Conversely, pro-apoptotic signaling molecules associated with the UPR, including pASK1 and CHOP were both noted to be attenuated. This state of ER stress signaling is likely contributive to the ability of Hodgkin cells to survive in a chronic ER stress environment from accumulation of viral proteins, as Hodgkin cells are closely associated with the Epstein-Barr virus [36].
Furthermore, a more recent study of tyrosine kinase inhibitors (TKIs) on Hodgkin cells has demonstrated an increase in ER stress and ER stress-induced apoptosis. L-428, L-1236, and KM-H2 cells were treated in vitro with the TKI sorafenib and monitored for markers of the UPR. Phosphorylation of PERK was noted, with a corresponding decrease in total intracellular signaling attributed to global protein translation inhibition via phosphorylation of eIF2α. In addition, pro-apoptotic signaling molecules GADD34 and CHOP were noted to be upregulated after incubation with sorafenib [37].
The ER stress response is clearly important to the survival of Hodgkin cells, and it has been shown that the UPR can likely be exploited through therapeutic means for treatment. The question remains as to whether the mechanism of action in proteasome inhibitors in Hodgkin disease is related to ER stress and the UPR, and if so, whether the increased UPR is responsible for radiosensitization. It is known that XBP1 is upregulated in these cells, and a downstream product of XBP1 is EDEM, a critical protein for the targeting of misfolded glycoproteins for degradation by the proteasome. Does the inhibition of the proteasome result in an increased concentration of misfolded glycoproteins in the ER of Hodgkin cells, leading to a terminal ER stress response? Furthermore, the mechanistic behind the increased radiosensitivity of proteasome inhibitor treated Hodgkin cells could provide a basis for research into potential future chemoradiotherapeutic regimens.
Pancreatic Cancer
Most pancreatic cancers are pancreatic ductal adenocarcinomas (PDACs), those that arise from the epithelium of pancreatic ducts. These cancers have an extremely poor prognosis, with a 1 year relative survival of 20% according to the American Cancer Society. As diagnosis is often late in the disease process, it is often difficult or impossible to surgically resect with clear margins. In this clinical context, novel approaches to chemotherapeutic treatment have the potential to improve outcomes.
Pancreatic cancers have been shown to become significantly hypoxic as they grow, with large volumes of the tumor at only a small fraction at the oxygenation levels of the surrounding normal pancreas [38]. In these conditions, one would expect high levels of ER stress due not only to hypoxia, but likely glucose depletion as well. Cellular adaptation to this environment is certainly linked to the survival and proliferation of malignant cells. Choe et al. showed that in pancreatic cancer cells, activation of the PERK and IRE1 arms of the UPR are delayed in the presence of ER stressors, compared to normal pancreatic cells. This was attributed to an abundance of protein folding machinery, such as chaperones. Additionally, once activated, the pro-survival XBP1 was noted to be activated for a longer time period in cancer cells when compared to normal cells [39]. As well as adapting to chronic ER stress, it has been recently postulated that anterior-gradient 2 (AGR2) may contribute to the initiation and development of PDAC [40]. As AGR2 is a gene associated with protein folding and maturation which is expressed in pancreatic cells under ER stress conditions, it may become a key piece in our understanding of how PDACs arise, as well as a potential future therapeutic target.
As adaptation, to an environment conducive to ER stress, is essential for survival and propagation of pancreatic cancer cells, ER stress and the UPR may provide a future target for therapeutics. In vitro studies of diindolylmethane derivatives have been shown to be similar in effect to thapsigargin in inducing ER stress, followed by subsequent apoptosis via death receptor 5 (DR5) through induction by CHOP [41]. Proteasome inhibitors have seen interest as a potential treatment for pancreatic cancer due to their unique effects. Bortezomib has been shown to increase GRP78, CHOP, and c-Jun NH2 terminal kinase (JNK) in L3.6pl pancreatic cancer cells, yet interestingly blocks PERK autophosphorylation, preventing phosphorylation of eIF2α and the subsequent translational inhibition [42, 43]. Without the suppression of translation, ubiquitinated proteins will accumulate, leading to a terminal ER stress response. More recently, a reovirus variant known as Reolysin has been shown to grow preferentially in cells with RAS activating mutations (found in about 90% of all pancreatic ductal adenocarcinomas). In pancreatic cancer cells, the virus causes an accumulation of viral protein products and accompanying ER stress and apoptosis. Adding bortezomib increases the rate of ER stress mediated apoptosis, as it would be expected after inhibiting a major pathway for protein degradation in an already stressed cell [44]. Treatments like these carry future hope for a disease which is commonly accompanied by a bleak prognosis.
Hepatocellular Carcinoma
Hepatocellular carcinoma (HCC) is a cancer of liver hepatocytes. In the United States, it is the second most lethal cancer after PDAC, with an 8.9% 5-year survival. Etiology is broad, with risk factors including hepatitis B and C viruses, chronic alcohol consumption, diabetes, non-alcoholic fatty liver disease, aflatoxin-B1, as well as a variety of other liver diseases [45]. Common to all the listed etiologies is the capacity to induce ER stress [46-53]. From the turn of the 21st century, hepatitis C has become the primary associated risk factor for HCC in the US, with approximately 60% seropositivity [54].
A study of human tissue infected with hepatitis C virus found morphological evidence of ER stress as a dilated and dysfunctional phenotype. Phosphorylated PERK, activated ATF6, and spliced XBP1 was increased compared to uninfected tissue, indicating that there was activation of the UPR response proteins, however, downstream UPR genes such as those encoding for XBP1, ATF6, GADD34, molecular chaperones, and calcium pump ATP2A2 were not induced. GRP78, however was found to be accumulated, suggesting a post-transcriptional mechanism of upregulation that may serve to increase protein folding capacity [55]. GRP78 is also found constitutively overexpressed in HCC cells, which may confer chemotherapeutic resistance [48].
As GRP78 appears to confer resistance to chemotherapy, it may be a future therapeutic target. The histone deacetylase inhibitor vorinostat was reported to acetylate GRP78, causing dissociation from, and subsequent activation of PERK [56]. In Huh7 HCC cells treated with vorinostat, phosphorylation of PERK and eIF2α was observed. Autophagy was induced, reportedly through downregulation of the Akt/mTOR pathway [57]. The UPR may be culpable, as ER stress has been shown to suppress the AKT/mTOR pathway [58].
As HCC is resistant to most chemotherapeutic agents, reducing this resistance may prove effective as a novel agent. Doxorubicin is widely used to treat HCC, although there is no clear survival benefit [59]. Doxorubicin resistance has been shown to be conferred through the UPR in multiple studies by pretreating with the known ER stress inducer tunicamycin [60, 61]. Researchers have found that by treating HepG2 HCC cells with the plant derived phenolic compound paeonol, doxorubicin resistance was abrogated by preventing ER stress induced activation of COX-2 and the downstream PI3K/Akt pathway [60]. Further research will be needed to determine if these pathways are effective in vivo, and ultimately in clinical cancer treatment.
Radiotherapy as an Inducer of ER Stress
Radiotherapy is accepted as a mainstay treatment against a wide variety of malignancies, including some of the most common cancers such as lung, breast, and prostate cancers. It has the advantage of being focal, such that the damage can be localized around the target tissue, avoiding systemic toxicity. Radiotherapy is not without risk however, as it can result in toxicity to any tissues in the beam path, and it can result in secondary malignancies. The classical understanding of the effects of radiotherapy centers around the generation of DNA single and double strand breaks either directly through DNA ionization or through interaction with generated reactive oxygen species. Unsuccessful repair of these breaks results in cell death [62]. A more complicated picture of the effects of radiotherapy has become apparent, with activation of multiple cell signaling cascades such as activation of membrane sphingomyelinase, activation of p53, activation of cell death receptors, and through induction of ER stress [63-65]. As ER stress plays a key role in cell survival as well as apoptosis, induction of ER stress or pharmacological manipulation of downstream UPR pathways may provide a key to improving the therapeutic ratio, resulting in increased tumor cell killing and reduction of toxicity to surrounding tissue.
Many studies have associated IR with ER stress and activation of the UPR in both cancer cells as well as normal cells [66-71]. In MCF-7 breast cancer cells, IR caused activation of the PERK- eIF2α pathway. This was related to cell death, as PERK knockdown through siRNA conferred survival [68]. In another study of MDA-MB-231 cancer cells, similar results were found, in that PERK knockdown conferred survival to IR. Expression of downstream proteins GRP78 and pro-apoptotic CHOP however, were not increased despite phosphorylation of eIF2α by PERK. This may suggest that IR induced ER stress acts in a CHOP independent manner [69]. Not all research indicates that PERK knockdown increases survival. Another group found that siRNA interference of PERK, ATF4, LAMP3 as well as pharmacological inhibition of PERK leads to decreased clonogenic survival to IR [72].
Research in normal tissues shows activation of the UPR in response to IR. Pulmonary artery endothelial cells were found to respond to IR doses of up to 50 Gy by activation of UPR signal transduction pathways associated with PERK and ATF6, although IRE1 splicing of XBP1 was not detected [66]. Normal rat intestinal epithelial (IEC-6 line) cells treated with 10 Gy of IR was reported to result in ER stress. UPR pathways associated with PERK and IRE1 were activated, including detection of XBP1 splicing, but the ATF6 pathway was not involved [67]. In human vascular endothelial cells, only the PERK- eIF2α pathway was activated in response to IR. These researchers postulated that ER stress may not be induced in this cell type, rather the PERK arm of the UPR may be activated due to interaction with lipid peroxidation products in the ER membrane (Figure 3) [70].
A hypothetical model of IR induced ER stress and activation of the UPR. IR generated reactive oxygen products react with lipids in the ER membrane. These peroxidation products activate the PERK arm of the UPR. If a strong enough ER stress response is invoked, the balance will be tipped towards apoptosis via the pro-apoptotic UPR genes.
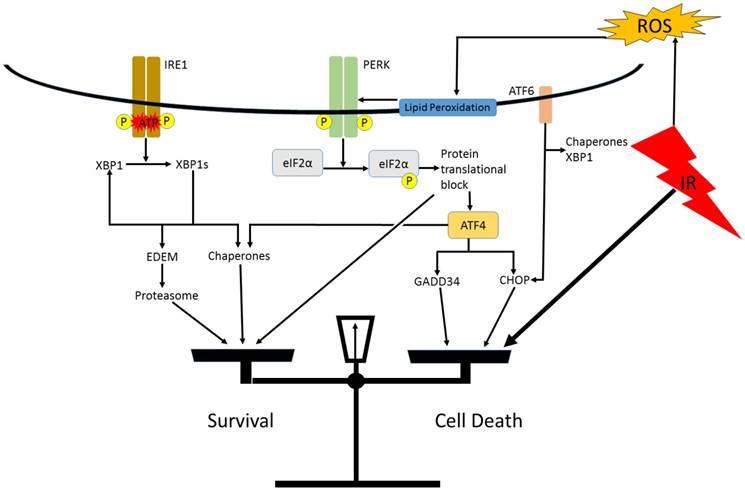
Current literature suggests that IR can result in ER stress and/or activation of the UPR in at least certain cell types in vitro, however, research has not yet elucidated the mechanism by which this occurs. Zhang et al. postulates that reactive oxygen species resulting from IR activates ER membrane receptors associated with the UPR [67]. This theory is consistent with other research showing ER stress induction by chemical induction of reactive oxygen species (ROS) [73, 74]. There has been little in vivo research on ER stress human subjects after radiotherapy, but one recent study found evidence of UPR activation via a significant upregulation of UPR genes (an average of more than 4-fold increase) including ATF4, CHOP, XBP1, GRP78, LC3, and ATG5 in peripheral lymphocytes after exposure to as little as 1 Gy when compared to non-irradiated control subjects [75]. Further research is necessary to determine how IR activates UPR effectors, by what mechanism, and whether it is dependent on cell type, or another undiscovered factor.
As the UPR clearly plays a role in in the response to radiotherapy, pharmacological manipulation of the UPR or induction of ER stress could be utilized to radiosensitize cancer cells. As cancer cells may rely on the UPR more heavily for survival, this type of treatment could preferentially affect cancer cells while sparing normal cells. The most basic method would be administration of an agent known to invoke ER stress, followed by administration of radiotherapy. The HIV protease inhibitor, nelfinavir, has been shown to inhibit the proteasome. As the proteasome is critical for degradation of misfolded proteins, inhibition is hypothesized to result in the buildup of misfolded proteins in the ER causing ER stress. After administration of nelfinavir to SQ20B head and neck cancer cells, phosphorylation of PERK and eIF2α was noted, along with increased expression of the downstream protein GADD34. These treated cells showed decreased surviving fraction on clonogenic assay after administration of IR, indicating radiosensitization [76]. Research on the proteasome inhibitor, bortezomib, has shown similar radiosensitization, although they have focused on pathways other than the UPR [77]. Other inducers of ER stress such as celecoxib and monascuspiloin have been demonstrated as radiosensitizers in vitro [78, 79].
A recent novel research pharmaceutical, GSK2606414, has been identified to selectively inhibit PERK, which could act as a radiosensitizer. It has been shown to be orally bioavailable, and inhibit tumor growth in vivo [80]. When combined with exposure to radiotherapy in glioblastoma cells, a 70-80% reduction in the genes downstream PERK was noted [81]. Further radiobiology research should certainly explore the effect of this drug along with radiation on various cancer cell lines. Future drugs like this could elucidate the mechanism of radiation resistance and possibly lead to a potent radiosensitizing therapeutic regimen.
Radiotherapy, along with surgery and chemotherapy make up the classic triad in the treatment of cancer. When these fail due to lack of efficacy or excessive side effects, there is the promise of the rapidly growing field of immunotherapy, made up of treatments such as interferons, cytokines, monoclonal antibodies, checkpoint inhibitors, dendritic cell therapy, T-cell engaging antibodies, and autologous vaccines.
Immune Modulation by ER Stress
ER stress is becoming recognized as having a role in the fundamental state of immune system, allowing and often promoting propagation of cancer cells. Furthermore, ER stress is recognized as playing an integral role, in cancer cells evading the immune system, through dysfunction in professional antigen presenting cells (APC), or modulation of antigen presentation through major histocompatibility complex (MHC) molecules. Research into these pathways may lead to future cancer immunotherapies that allow the immune system to identify these cancer cells.
Besides its role in the survival of tumor cells which have a large burden of misfolded proteins, the UPR is involved in producing the inflammatory microenvironment conducive to tumor growth. Cancer cells under ER stress have been shown to transmit ER stress to surrounding macrophages, causing upregulation of GRP78, GADD34, CHOP, and spliced XBP1 [82]. These macrophages in turn release pro-inflammatory cytokines, chemokines, and metalloproteases. Two specific secreted cytokines, IL-6 and IL-23, have been implicated in tumor growth [83, 84]. Dendritic cells (DCs) are also responsive to transmitted ER stress in the tumor environment, releasing not only pro-inflammatory, pro-tumorigenic cytokines in response to transmitted ER stress, but also arginase, which effects T cells in a suppressive manner [85]. DCs have also been shown to increase IL-23 transcription and secretion upon the induction of ER stress, with CHOP as the responsible transcription factor, thus linking the UPR to specific DC released tumor growth factors [86]. Even in a pro-tumorigenic environment, cancer cells must evade the immune system to proliferate.
Antigen presentation is key to the immune system's ability to mount an effective response to malignant cells. Antigens are presented on cell surface MHC class I from a sampling of cytosolic and ER contents. As ER stress changes the distribution of proteins in the cytosol and ER via PERK-mediated protein translational attenuation, and causes upregulation of molecular chaperones and protein degradation via the ERAD system and proteasome, it would be expected that the UPR would influence antigen presentation. Evidence suggests that epitopes derived from defective proteins are preferentially presented on MHC class I molecules, likely due to increased degradation rate of these problem proteins [87, 88]. This does not necessarily translate into increased presentation of antigens on the surface of cells undergoing ER stress. Cells under ER stress have been shown to have decreased MHC class I antigen presentation, hypothetically believed to be due to global protein translational attenuation [89]. Additionally, the role of a micro-RNA, miRNA-346, has been implicated in reduced antigen presentation of cells under ER stress. Spliced XBP1 induces miRNA-346, resulting in a decrease in antigen presentation through attenuation of MHC class I, interferon induced genes, and transporter associated with antigen processing 1 (TAP1) [90]. This pathway can be connected to human malignancy, as miRNA-346 has been identified to be significantly overexpressed in follicular thyroid carcinoma [91].
DCs can be looked upon as the master controllers of adaptive immunity, and are necessary for T-cell dependent immunity against cancer cells. UPR associated protein XBP1 has been reported to be constitutively active in DCs, and identified as necessary for development and differentiation [92, 93]. IRE1, upstream of XBP1 has been implicated in the control of DC antigen processing and cell to cell contact in addition to maintaining ER homeostasis [93]. Some tumor types, such as ovarian carcinoma, can suppress dendritic cell mediated T cell immunity through a type of dendritic cell dysfunction. Lipid peroxide products lead to chronic ER stress in DCs, causing a high constitutive activation of XPB1, leading to triglyceride biosynthesis and accumulation [94]. This causes a dysfunction that can be alleviated through the suppression of XBP1, restoring the anti-tumor immune response. One might wonder if immune function could be restored by pharmacological inhibition of IRE1, such as through the proteasome inhibitor, bortezomib, which has been shown to suppress IRE1 and subsequent XBP1 splicing [95].
Bringing therapies together for multi-directional attack. Potential therapeutic combination of immunotherapy and inducers of ER stress (radiation, or anthracyclines). These ER stressors induce the expression of calreticulin on the surface of tumor cells. This acts as a signal for phagocytosis of injured cells by dendritic cells, and subsequent activation of cell mediated immunity. Cells expression calreticulin from ER stress inducers such as ionizing radiation have been shown to have increased sensitivity to cytotoxic T-lymphocyte lysis123.
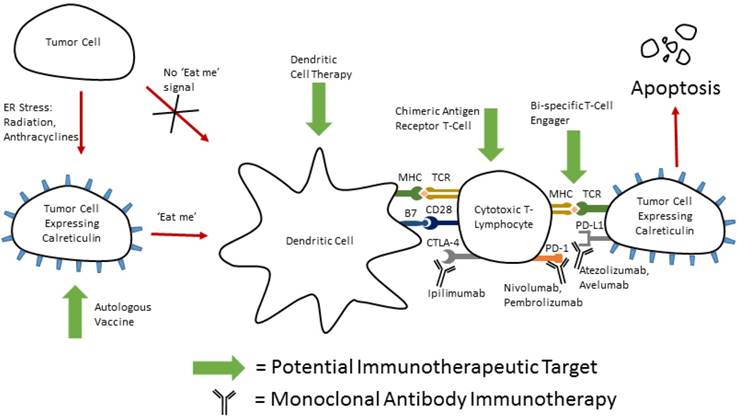
Functional DCs can lead to activation of cytotoxic T lymphocytes to mount an immune response to cancer cells under the correct conditions. Before they can activate these T cells, they must internalize antigen associated with the target cells. Calreticulin (CRT), an ER resident molecular chaperone has been shown to function as an “eat me” signal to DCs when exposed on the cell surface. CRT is presented on the cell surface after induction of ER stress in a PERK dependent fashion, reliant upon phosphorylation of eIF2α [96-98]. Agents capable of inducing CRT exposure include gamma radiation, UVC light, and chemotherapeutic agents such as anthracyclines, and oxaliplatin [96, 99, 100]. Recent research hints that CRT may have a role in clinical oncology. In human patients with non-small cell lung cancer, there was a strong correlation between CRT expression on the surface of tumor cells, and not only an increase in local DC and effector memory T-cells, but a better prognosis as well [101]. It is not understood, however, why some of these tumors have low CRT expression or whether treatment could be directed to increase expression. It is therefore a relevant question to ask whether we can invoke ER stress therapeutically to cause CRT expression and trigger immunogenic cell death. A fascinating potential therapy could be realized by combining inducers of CRT and antigen presentation with immunotherapy or cancer vaccines (Figure 4).
Conclusion
The UPR is critically important for the survival of cells exposed to conditions of ER stress. Due to local tumor microenvironments, coupled with high protein synthesis demands, cancer cells are uniquely reliant on the UPR for survival in a way that normal cells are not. Future therapeutic targets could target specific UPR pathways that cancer cells rely upon, while leaving normal cells relatively intact. In cancer cells under chronic ER stress, therapy could be targeted to tip the balance towards apoptosis through the induction of additional ER stress through chemotherapy, radiotherapy as well as through more exotic means such as engineered viruses. An essential question is how the normal tissues will survive in the presence of ER stress inducing treatment. It may be that without the existing insult of oxygen and nutrient starvation, upregulated UPR machinery and high protein demand, that ER stress is maintained at sub-lethal levels in comparison with rapidly dividing malignant cells. Future research should not only illuminate whether ER stress and the UPR can be harnessed against malignancies, but also elucidate the consequence to normal tissues of an insult great enough to drive malignant cells to a terminal ER stress response.
Acknowledgements
This work was supported by grants from NIDDK K01DK096032 (S.S.).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Bertolotti A, Zhang Y, Hendershot LM, Harding HP, Ron D. Dynamic interaction of BiP and ER stress transducers in the unfolded-protein response. Nature cell biology. 2000;2:326-32
2. Shen J, Snapp EL, Lippincott-Schwartz J, Prywes R. Stable binding of ATF6 to BiP in the endoplasmic reticulum stress response. Mol Cell Biol. 2005;25:921-32
3. Harding HP, Zhang Y, Ron D. Protein translation and folding are coupled by an endoplasmic-reticulum-resident kinase. Nature. 1999;397:271-4
4. Koumenis C, Naczki C, Koritzinsky M, Rastani S, Diehl A, Sonenberg N. et al. Regulation of Protein Synthesis by Hypoxia via Activation of the Endoplasmic Reticulum Kinase PERK and Phosphorylation of the Translation Initiation Factor eIF2. Molecular and Cellular Biology. 2002;22:7405-16
5. Blais JD, Filipenko V, Bi M, Harding HP, Ron D, Koumenis C. et al. Activating transcription factor 4 is translationally regulated by hypoxic stress. Mol Cell Biol. 2004;24:7469-82
6. Harding HP, Novoa I, Zhang Y, Zeng H, Wek R, Schapira M. et al. Regulated Translation Initiation Controls Stress-Induced Gene Expression in Mammalian Cells. Molecular Cell. 2000;6:1099-108
7. Oyadomari S, Mori M. Roles of CHOP/GADD153 in endoplasmic reticulum stress. Cell Death & Differentiation. 2004;11:381-9
8. Luo S, Baumeister P, Yang S, Abcouwer SF, Lee AS. Induction of Grp78/BiP by translational block: activation of the Grp78 promoter by ATF4 through and upstream ATF/CRE site independent of the endoplasmic reticulum stress elements. J Biol Chem. 2003;278:37375-85
9. Brush MH, Weiser DC, Shenolikar S. Growth Arrest and DNA Damage-Inducible Protein GADD34 Targets Protein Phosphatase 1 to the Endoplasmic Reticulum and Promotes Dephosphorylation of the Subunit of Eukaryotic Translation Initiation Factor 2. Molecular and Cellular Biology. 2003;23:1292-303
10. Maytin EV, Ubeda M, Lin JC, Habener JF. Stress-inducible transcription factor CHOP/gadd153 induces apoptosis in mammalian cells via p38 kinase-dependent and -independent mechanisms. Exp Cell Res. 2001;267:193-204
11. Chen X, Shen J, Prywes R. The luminal domain of ATF6 senses endoplasmic reticulum (ER) stress and causes translocation of ATF6 from the ER to the Golgi. J Biol Chem. 2002;277:13045-52
12. Yoshida H, Haze K, Yanagi H, Yura T, Mori K. Identification of the cis-acting endoplasmic reticulum stress response element responsible for transcriptional induction of mammalian glucose-regulated proteins Involvement of basic leucine zipper transcription factors. Journal of Biological Chemistry. 1998;273:33741-9
13. Yoshida H, Okada T, Haze K, Yanagi H, Yura T, Negishi M. et al. ATF6 activated by proteolysis binds in the presence of NF-Y (CBF) directly to the cis-acting element responsible for the mammalian unfolded protein response. Molecular and cellular biology. 2000;20:6755-67
14. Yoshida H, Matsui T, Yamamoto A, Okada T, Mori K. XBP1 mRNA is induced by ATF6 and spliced by IRE1 in response to ER stress to produce a highly active transcription factor. Cell. 2001;107:881-91
15. Lee K, Tirasophon W, Shen X, Michalak M, Prywes R, Okada T. et al. IRE1-mediated unconventional mRNA splicing and S2P-mediated ATF6 cleavage merge to regulate XBP1 in signaling the unfolded protein response. Genes & development. 2002;16:452-66
16. Hosokawa N, Wada I, Hasegawa K, Yorihuzi T, Tremblay LO, Herscovics A. et al. A novel ER α-mannosidase-like protein accelerates ER-associated degradation. EMBO reports. 2001;2:415-22
17. Oda Y, Hosokawa N, Wada I, Nagata K. EDEM as an acceptor of terminally misfolded glycoproteins released from calnexin. Science. 2003;299:1394-7
18. Yoshida H, Matsui T, Hosokawa N, Kaufman RJ, Nagata K, Mori K. A time-dependent phase shift in the mammalian unfolded protein response. Developmental cell. 2003;4:265-71
19. Calfon M, Zeng H, Urano F, Till JH, Hubbard SR, Harding HP. et al. IRE1 couples endoplasmic reticulum load to secretory capacity by processing the XBP-1 mRNA. Nature. 2002;415:92-6
20. Nishitoh H, Saitoh M, Mochida Y, Takeda K, Nakano H, Rothe M. et al. ASK1 is essential for JNK/SAPK activation by TRAF2. Molecular cell. 1998;2:389-95
21. Urano F, Wang X, Bertolotti A, Zhang Y, Chung P, Harding HP. et al. Coupling of stress in the ER to activation of JNK protein kinases by transmembrane protein kinase IRE1. Science. 2000;287:664-6
22. Kato H, Nakajima S, Saito Y, Takahashi S, Katoh R, Kitamura M. mTORC1 serves ER stress-triggered apoptosis via selective activation of the IRE1-JNK pathway. Cell Death Differ. 2012;19:310-20
23. Yu Z, Pestell TG, Lisanti MP, Pestell RG. Cancer stem cells. Int J Biochem Cell Biol. 2012;44:2144-51
24. Luo S, Mao C, Lee B, Lee AS. GRP78/BiP is required for cell proliferation and protecting the inner cell mass from apoptosis during early mouse embryonic development. Mol Cell Biol. 2006;26:5688-97
25. Miharada K, Karlsson G, Rehn M, Rorby E, Siva K, Cammenga J. et al. Cripto regulates hematopoietic stem cells as a hypoxic-niche-related factor through cell surface receptor GRP78. Cell Stem Cell. 2011;9:330-44
26. Bartkowiak K, Effenberger KE, Harder Sn, Andreas A, Buck F, Peter-Katalinic J. et al. Discovery of a novel unfolded protein response phenotype of cancer stem/progenitor cells from the bone marrow of breast cancer patients. Journal of proteome research. 2010;9:3158-68
27. Li B, Cheng XL, Yang YP, Li ZQ. GRP78 mediates radiation resistance of a stem cell-like subpopulation within the MCF-7 breast cancer cell line. Oncol Rep. 2013;30:2119-26
28. Fu Y, Wey S, Wang M, Ye R, Liao CP, Roy-Burman P. et al. Pten null prostate tumorigenesis and AKT activation are blocked by targeted knockout of ER chaperone GRP78/BiP in prostate epithelium. Proc Natl Acad Sci U S A. 2008;105:19444-9
29. Daneshmand S, Quek ML, Lin E, Lee C, Cote RJ, Hawes D. et al. Glucose-regulated protein GRP78 is up-regulated in prostate cancer and correlates with recurrence and survival. Hum Pathol. 2007;38:1547-52
30. Hill RP, Marie-Egyptienne DT, Hedley DW. Cancer stem cells, hypoxia and metastasis. Semin Radiat Oncol. 2009;19:106-11
31. Wu M-J, Jan C-I, Tsay Y-G, Yu Y-H, Huang C-Y, Lin S-C. et al. Elimination of head and neck cancer initiating cells through targeting glucose regulated protein78 signaling. Molecular cancer. 2010;9:1
32. Dadey DY, Kapoor V, Hoye K, Khudanyan A, Collins A, Thotala D. et al. Antibody targeting GRP78 enhances the efficacy of radiation therapy in human glioblastoma and non-small-cell lung cancer cell lines and tumor models. Clinical Cancer Research. 2016: clincanres. 1935:2016
33. Ferrara F, Staquicini DI, Driessen WH, D'Angelo S, Dobroff AS, Barry M. et al. Targeted molecular-genetic imaging and ligand-directed therapy in aggressive variant prostate cancer. Proceedings of the National Academy of Sciences. 2016:201615400
34. van Galen P, Kreso A, Mbong N, Kent DG, Fitzmaurice T, Chambers JE. et al. The unfolded protein response governs integrity of the haematopoietic stem-cell pool during stress. Nature. 2014;510:268-72
35. Pajonk F, Pajonk K, McBride WH. Apoptosis and radiosensitization of Hodgkin cells by proteasome inhibition. International Journal of Radiation Oncology* Biology* Physics. 2000;47:1025-32
36. Chang KC, Chen PC, Chen YP, Chang Y, Su IJ. Dominant expression of survival signals of endoplasmic reticulum stress response in Hodgkin lymphoma. Cancer Sci. 2011;102:275-81
37. Holz MS, Janning A, Renne C, Gattenlohner S, Spieker T, Brauninger A. Induction of endoplasmic reticulum stress by sorafenib and activation of NF-kappaB by lestaurtinib as a novel resistance mechanism in Hodgkin lymphoma cell lines. Mol Cancer Ther. 2013;12:173-83
38. Koong AC, Mehta VK, Le QT, Fisher GA, Terris DJ, Brown JM. et al. Pancreatic tumors show high levels of hypoxia. International Journal of Radiation Oncology* Biology* Physics. 2000;48:919-22
39. Choe JH. Altered Responses to Endoplasmic Reticulum Stress in Pancreatic Cancer. 2011.
40. Dumartin L, Alrawashdeh W, Trabulo SM, Radon TP, Steiger K, Feakins RM. et al. ER stress protein AGR2 precedes and is involved in the regulation of pancreatic cancer initiation. Oncogene. 2016
41. Abdelrahim M, Newman K, Vanderlaag K, Samudio I, Safe S. 3,3'-diindolylmethane (DIM) and its derivatives induce apoptosis in pancreatic cancer cells through endoplasmic reticulum stress-dependent upregulation of DR5. Carcinogenesis. 2006;27:717-28
42. Nawrocki ST, Carew JS, Dunner K Jr, Boise LH, Chiao PJ, Huang P. et al. Bortezomib inhibits PKR-like endoplasmic reticulum (ER) kinase and induces apoptosis via ER stress in human pancreatic cancer cells. Cancer Res. 2005;65:11510-9
43. Nawrocki ST, Carew JS, Pino MS, Highshaw RA, Dunner K Jr, Huang P. et al. Bortezomib sensitizes pancreatic cancer cells to endoplasmic reticulum stress-mediated apoptosis. Cancer Res. 2005;65:11658-66
44. Carew JS, Espitia CM, Zhao W, Kelly KR, Coffey M, Freeman JW. et al. Reolysin is a novel reovirus-based agent that induces endoplasmic reticular stress-mediated apoptosis in pancreatic cancer. Cell Death Dis. 2013;4:e728
45. Farazi PA, DePinho RA. Hepatocellular carcinoma pathogenesis: from genes to environment. Nat Rev Cancer. 2006;6:674-87
46. Petrasek J, Iracheta-Vellve A, Csak T, Satishchandran A, Kodys K, Kurt-Jones EA. et al. STING-IRF3 pathway links endoplasmic reticulum stress with hepatocyte apoptosis in early alcoholic liver disease. Proceedings of the National Academy of Sciences. 2013;110:16544-9
47. Paul S, Jakhar R, Bhardwaj M, Kang SC. Glutathione-S-transferase omega 1 (GSTO1-1) acts as mediator of signaling pathways involved in aflatoxin B1-induced apoptosis-autophagy crosstalk in macrophages. Free Radic Biol Med. 2015;89:1218-30
48. Shuda M, Kondoh N, Imazeki N, Tanaka K, Okada T, Mori K. et al. Activation of the ATF6, XBP1 and grp78 genes in human hepatocellular carcinoma: a possible involvement of the ER stress pathway in hepatocarcinogenesis. Journal of Hepatology. 2003;38:605-14
49. Pavio N, Romano PR, Graczyk TM, Feinstone SM, Taylor DR. Protein Synthesis and Endoplasmic Reticulum Stress Can Be Modulated by the Hepatitis C Virus Envelope Protein E2 through the Eukaryotic Initiation Factor 2 Kinase PERK. Journal of Virology. 2003;77:3578-85
50. Hsieh YH, Su IJ, Wang HC, Chang WW, Lei HY, Lai MD. et al. Pre-S mutant surface antigens in chronic hepatitis B virus infection induce oxidative stress and DNA damage. Carcinogenesis. 2004;25:2023-32
51. Wei Y, Wang D, Topczewski F, Pagliassotti MJ. Saturated fatty acids induce endoplasmic reticulum stress and apoptosis independently of ceramide in liver cells. Am J Physiol Endocrinol Metab. 2006;291:E275-81
52. Stickel F, Hellerbrand C. Non-alcoholic fatty liver disease as a risk factor for hepatocellular carcinoma: mechanisms and implications. Gut. 2010;59:1303-7
53. Zhang Q, Li Y, Liang T, Lu X, Zhang C, Liu X. et al. ER stress and autophagy dysfunction contribute to fatty liver in diabetic mice. Int J Biol Sci. 2015;11:559-68
54. de Martel C, Maucort-Boulch D, Plummer M, Franceschi S. World-wide relative contribution of hepatitis B and C viruses in hepatocellular carcinoma. Hepatology. 2015;62:1190-200
55. Asselah T, Bieche I, Mansouri A, Laurendeau I, Cazals-Hatem D, Feldmann G. et al. In vivo hepatic endoplasmic reticulum stress in patients with chronic hepatitis C. J Pathol. 2010;221:264-74
56. Kahali S, Sarcar B, Fang B, Williams ES, Koomen JM, Tofilon PJ. et al. Activation of the Unfolded Protein Response Contributes toward the Antitumor Activity of Vorinostat. Neoplasia. 2010;12:80-6
57. Liu Y-L, Yang P-M, Shun C-T, Wu M-S, Weng J-R, Chen C-C. Autophagy potentiates the anti-cancer effects of the histone deacetylase inhibitors in hepatocellular carcinoma. Autophagy. 2014;6:1057-65
58. Qin L, Wang Z, Tao L, Wang Y. ER stress negatively regulates AKT/TSC/mTOR pathway to enhance autophagy. Autophagy. 2014;6:239-47
59. Abou-Alfa GK, Johnson P, Knox JJ, Capanu M, Davidenko I, Lacava J. et al. Doxorubicin plus sorafenib vs doxorubicin alone in patients with advanced hepatocellular carcinoma: a randomized trial. Jama. 2010;304:2154-60
60. Fan L, Song B, Sun G, Ma T, Zhong F, Wei W. Endoplasmic reticulum stress-induced resistance to doxorubicin is reversed by paeonol treatment in human hepatocellular carcinoma cells. PLoS One. 2013;8:e62627
61. Fan L, Sun G, Ma T, Zhong F, Lei Y, Li X. et al. Melatonin reverses tunicamycin-induced endoplasmic reticulum stress in human hepatocellular carcinoma cells and improves cytotoxic response to doxorubicin by increasing CHOP and decreasing Survivin. Journal of pineal research. 2013;55:184-94
62. Ross G. Induction of cell death by radiotherapy. Endocrine-related cancer. 1999;6:41-4
63. Moretti L, Cha YI, Niermann KJ, Lu B. Switch between apoptosis and autophagy: radiation-induced endoplasmic reticulum stress? Cell Cycle. 2007;6:793-8
64. Gudkov AV, Komarova EA. The role of p53 in determining sensitivity to radiotherapy. Nat Rev Cancer. 2003;3:117-29
65. Dent P, Yacoub A, Contessa J, Caron R, Amorino G, Valerie K. et al. Stress and Radiation-Induced Activation of Multiple Intracellular Signaling Pathways1. Radiation Research. 2003;159:283-300
66. Panganiban RA, Mungunsukh O, Day RM. X-irradiation induces ER stress, apoptosis, and senescence in pulmonary artery endothelial cells. Int J Radiat Biol. 2013;89:656-67
67. Zhang B, Wang Y, Pang X, Su Y, Ai G, Wang T. ER stress induced by ionising radiation in IEC-6 cells. Int J Radiat Biol. 2010;86:429-35
68. Kim KW, Moretti L, Mitchell LR, Jung DK, Lu B. Endoplasmic reticulum stress mediates radiation-induced autophagy by perk-eIF2alpha in caspase-3/7-deficient cells. Oncogene. 2010;29:3241-51
69. Oommen D, Prise KM. Down-regulation of PERK enhances resistance to ionizing radiation. Biochem Biophys Res Commun. 2013;441:31-5
70. Kim EJ, Lee YJ, Kang S, Lim YB. Ionizing radiation activates PERK/eIF2alpha/ATF4 signaling via ER stress-independent pathway in human vascular endothelial cells. Int J Radiat Biol. 2014;90:306-12
71. Li F, Zheng X, Liu Y, Li P, Liu X, Ye F. et al. Different Roles of CHOP and JNK in Mediating Radiation-Induced Autophagy and Apoptosis in Breast Cancer Cells. Radiat Res. 2016;185:539-48
72. Nagelkerke A, Bussink J, van der Kogel AJ, Sweep FC, Span PN. The PERK/ATF4/LAMP3-arm of the unfolded protein response affects radioresistance by interfering with the DNA damage response. Radiother Oncol. 2013;108:415-21
73. Watanabe Y, Suzuki O, Haruyama T, Akaike T. Interferon-gamma induces reactive oxygen species and endoplasmic reticulum stress at the hepatic apoptosis. J Cell Biochem. 2003;89:244-53
74. Xue X, Piao JH, Nakajima A, Sakon-Komazawa S, Kojima Y, Mori K. et al. Tumor necrosis factor alpha (TNFalpha) induces the unfolded protein response (UPR) in a reactive oxygen species (ROS)-dependent fashion, and the UPR counteracts ROS accumulation by TNFalpha. J Biol Chem. 2005;280:33917-25
75. Saglar E, Unlu S, Babalioglu I, Gokce SC, Mergen H. Assessment of ER Stress and autophagy induced by ionizing radiation in both radiotherapy patients and ex vivo irradiated samples. J Biochem Mol Toxicol. 2014;28:413-7
76. Gupta AK, Li B, Cerniglia GJ, Ahmed MS, Hahn SM, Maity A. The HIV Protease Inhibitor Nelfinavir Downregulates Akt Phosphorylation by Inhibiting Proteasomal Activity and Inducing the Unfolded Protein Response. Neoplasia. 2007;9:271-8
77. Tamatani T, Takamaru N, Hara K, Kinouchi M, Kuribayashi N, Ohe G. et al. Bortezomib-enhanced radiosensitization through the suppression of radiation-induced nuclear factor-kappaB activity in human oral cancer cells. Int J Oncol. 2013;42:935-44
78. Chiu HW, Fang WH, Chen YL, Wu MD, Yuan GF, Ho SY. et al. Monascuspiloin enhances the radiation sensitivity of human prostate cancer cells by stimulating endoplasmic reticulum stress and inducing autophagy. PLoS One. 2012;7:e40462
79. Suzuki K, Gerelchuluun A, Hong Z, Sun L, Zenkoh J, Moritake T. et al. Celecoxib enhances radiosensitivity of hypoxic glioblastoma cells through endoplasmic reticulum stress. Neuro Oncol. 2013;15:1186-99
80. Axten JM, Medina JR, Feng Y, Shu A, Romeril SP, Grant SW. et al. Discovery of 7-methyl-5-(1-{[3-(trifluoromethyl)phenyl]acetyl}-2,3-dihydro-1H-indol-5-yl)-7H-p yrrolo[2,3-d]pyrimidin-4-amine (GSK2606414), a potent and selective first-in-class inhibitor of protein kinase R (PKR)-like endoplasmic reticulum kinase (PERK). J Med Chem. 2012;55:7193-207
81. Dadey D, Kapoor V, Khudanyan A, Thotala D, Hallahan D. Abstract 3323: Radiation-induced ER stress contributes to survival in glioblastoma cell lines. Cancer Research. 2015;75:3323
82. Mahadevan NR, Rodvold J, Sepulveda H, Rossi S, Drew AF, Zanetti M. Transmission of endoplasmic reticulum stress and pro-inflammation from tumor cells to myeloid cells. Proc Natl Acad Sci U S A. 2011;108:6561-6
83. Langowski JL, Zhang X, Wu L, Mattson JD, Chen T, Smith K. et al. IL-23 promotes tumour incidence and growth. Nature. 2006;442:461-5
84. Schafer ZT, Brugge JS. IL-6 involvement in epithelial cancers. J Clin Invest. 2007;117:3660-3
85. Mahadevan NR, Anufreichik V, Rodvold JJ, Chiu KT, Sepulveda H, Zanetti M. Cell-extrinsic effects of tumor ER stress imprint myeloid dendritic cells and impair CD8(+) T cell priming. PLoS One. 2012;7:e51845
86. Goodall JC, Wu C, Zhang Y, McNeill L, Ellis L, Saudek V. et al. Endoplasmic reticulum stress-induced transcription factor, CHOP, is crucial for dendritic cell IL-23 expression. Proceedings of the national academy of Sciences. 2010;107:17698-703
87. Golovina TN, Morrison SE, Eisenlohr LC. The Impact of Misfolding versus Targeted Degradation on the Efficiency of the MHC Class I-Restricted Antigen Processing. The Journal of Immunology. 2005;174:2763-9
88. Ostankovitch M, Robila V, Engelhard VH. Regulated Folding of Tyrosinase in the Endoplasmic Reticulum Demonstrates That Misfolded Full-Length Proteins Are Efficient Substrates for Class I Processing and Presentation. The Journal of Immunology. 2005;174:2544-51
89. Granados DP, Tanguay PL, Hardy MP, Caron E, de Verteuil D, Meloche S. et al. ER stress affects processing of MHC class I-associated peptides. BMC Immunol. 2009;10:10
90. Bartoszewski R, Brewer JW, Rab A, Crossman DK, Bartoszewska S, Kapoor N. et al. The unfolded protein response (UPR)-activated transcription factor X-box-binding protein 1 (XBP1) induces microRNA-346 expression that targets the human antigen peptide transporter 1 (TAP1) mRNA and governs immune regulatory genes. J Biol Chem. 2011;286:41862-70
91. Weber F, Teresi RE, Broelsch CE, Frilling A, Eng C. A limited set of human MicroRNA is deregulated in follicular thyroid carcinoma. J Clin Endocrinol Metab. 2006;91:3584-91
92. Iwakoshi NN, Pypaert M, Glimcher LH. The transcription factor XBP-1 is essential for the development and survival of dendritic cells. J Exp Med. 2007;204:2267-75
93. Osorio F, Tavernier SJ, Hoffmann E, Saeys Y, Martens L, Vetters J. et al. The unfolded-protein-response sensor IRE-1alpha regulates the function of CD8alpha+ dendritic cells. Nat Immunol. 2014;15:248-57
94. Cubillos-Ruiz JR, Silberman PC, Rutkowski MR, Chopra S, Perales-Puchalt A, Song M. et al. ER Stress Sensor XBP1 Controls Anti-tumor Immunity by Disrupting Dendritic Cell Homeostasis. Cell. 2015;161:1527-38
95. Hirai M, Kadowaki N, Kitawaki T, Fujita H, Takaori-Kondo A, Fukui R. et al. Bortezomib suppresses function and survival of plasmacytoid dendritic cells by targeting intracellular trafficking of Toll-like receptors and endoplasmic reticulum homeostasis. Blood. 2011;117:500-9
96. Obeid M, Tesniere A, Ghiringhelli F, Fimia GM, Apetoh L, Perfettini JL. et al. Calreticulin exposure dictates the immunogenicity of cancer cell death. Nat Med. 2007;13:54-61
97. Panaretakis T, Kepp O, Brockmeier U, Tesniere A, Bjorklund AC, Chapman DC. et al. Mechanisms of pre-apoptotic calreticulin exposure in immunogenic cell death. The EMBO journal. 2009;28:578-90
98. Panaretakis T, Joza N, Modjtahedi N, Tesniere A, Vitale I, Durchschlag M. et al. The co-translocation of ERp57 and calreticulin determines the immunogenicity of cell death. Cell Death Differ. 2008;15:1499-509
99. Obeid M, Panaretakis T, Joza N, Tufi R, Tesniere A, van Endert P. et al. Calreticulin exposure is required for the immunogenicity of gamma-irradiation and UVC light-induced apoptosis. Cell Death Differ. 2007;14:1848-50
100. Martins I, Kepp O, Schlemmer F, Adjemian S, Tailler M, Shen S. et al. Restoration of the immunogenicity of cisplatin-induced cancer cell death by endoplasmic reticulum stress. Oncogene. 2011;30:1147-58
101. Fucikova J, Becht E, Iribarren K, Goc J, Remark R, Damotte D. et al. Calreticulin Expression in Human Non-Small Cell Lung Cancers Correlates with Increased Accumulation of Antitumor Immune Cells and Favorable Prognosis. Cancer Res. 2016;76:1746-56
102. Pootrakul L, Datar RH, Shi SR, Cai J, Hawes D, Groshen SG. et al. Expression of stress response protein Grp78 is associated with the development of castration-resistant prostate cancer. Clin Cancer Res. 2006;12:5987-93
103. Sanchez AM, Martinez-Botas J, Malagarie-Cazenave S, Olea N, Vara D, Lasuncion MA. et al. Induction of the endoplasmic reticulum stress protein GADD153/CHOP by capsaicin in prostate PC-3 cells: a microarray study. Biochem Biophys Res Commun. 2008;372:785-91
104. Segawa T, Nau ME, Xu LL, Chilukuri RN, Makarem M, Zhang W. et al. Androgen-induced expression of endoplasmic reticulum (ER) stress response genes in prostate cancer cells. Oncogene. 2002;21:8749
105. Yamaguchi H, Wang HG. CHOP is involved in endoplasmic reticulum stress-induced apoptosis by enhancing DR5 expression in human carcinoma cells. J Biol Chem. 2004;279:45495-502
106. Bhutia SK, Dash R, Das SK, Azab B, Su ZZ, Lee SG. et al. Mechanism of autophagy to apoptosis switch triggered in prostate cancer cells by antitumor cytokine melanoma differentiation-associated gene 7/interleukin-24. Cancer Res. 2010;70:3667-76
107. Qu L, Huang S, Baltzis D, Rivas-Estilla AM, Pluquet O, Hatzoglou M. et al. Endoplasmic reticulum stress induces p53 cytoplasmic localization and prevents p53-dependent apoptosis by a pathway involving glycogen synthase kinase-3beta. Genes Dev. 2004;18:261-77
108. Yang JS, Chen GW, Hsia TC, Ho HC, Ho CC, Lin MW. et al. Diallyl disulfide induces apoptosis in human colon cancer cell line (COLO 205) through the induction of reactive oxygen species, endoplasmic reticulum stress, caspases casade and mitochondrial-dependent pathways. Food Chem Toxicol. 2009;47:171-9
109. Gandin V, Pellei M, Tisato F, Porchia M, Santini C, Marzano C. A novel copper complex induces paraptosis in colon cancer cells via the activation of ER stress signalling. J Cell Mol Med. 2012;16:142-51
110. Lu JJ, Chen SM, Zhang XW, Ding J, Meng LH. The anti-cancer activity of dihydroartemisinin is associated with induction of iron-dependent endoplasmic reticulum stress in colorectal carcinoma HCT116 cells. Invest New Drugs. 2011;29:1276-83
111. Pan MH, Hsieh MC, Kuo JM, Lai CS, Wu H, Sang S. et al. 6-Shogaol induces apoptosis in human colorectal carcinoma cells via ROS production, caspase activation, and GADD 153 expression. Mol Nutr Food Res. 2008;52:527-37
112. Uramoto H, Sugio K, Oyama T, Nakata S, Ono K, Yoshimastu T. et al. Expression of endoplasmic reticulum molecular chaperone Grp78 in human lung cancer and its clinical significance. Lung Cancer. 2005;49:55-62
113. Kim KM, Yu TK, Chu HH, Park HS, Jang KY, Moon WS. et al. Expression of ER stress and autophagy-related molecules in human non-small cell lung cancer and premalignant lesions. Int J Cancer. 2012;131:E362-70
114. Ling YH, Li T, Perez-Soler R, Haigentz M Jr. Activation of ER stress and inhibition of EGFR N-glycosylation by tunicamycin enhances susceptibility of human non-small cell lung cancer cells to erlotinib. Cancer Chemother Pharmacol. 2009;64:539-48
115. Hung JY, Hsu YL, Ni WC, Tsai YM, Yang CJ, Kuo PL. et al. Oxidative and endoplasmic reticulum stress signaling are involved in dehydrocostuslactone-mediated apoptosis in human non-small cell lung cancer cells. Lung Cancer. 2010;68:355-65
116. Nozaki S, Sledge Jr GW, Nakshatri H. Repression of GADD153/CHOP by NF-[kappa] B: a possible cellular defense against endoplasmic reticulum stress-induced cell death. Oncogene. 2001;20:2178
117. Fu Y, Li J, Lee AS. GRP78/BiP inhibits endoplasmic reticulum BIK and protects human breast cancer cells against estrogen starvation-induced apoptosis. Cancer Res. 2007;67:3734-40
118. Milani M, Rzymski T, Mellor HR, Pike L, Bottini A, Generali D. et al. The role of ATF4 stabilization and autophagy in resistance of breast cancer cells treated with Bortezomib. Cancer Res. 2009;69:4415-23
119. Ariazi EA, Cunliffe HE, Lewis-Wambi JS, Slifker MJ, Willis AL, Ramos P. et al. Estrogen induces apoptosis in estrogen deprivation-resistant breast cancer through stress responses as identified by global gene expression across time. Proc Natl Acad Sci U S A. 2011;108:18879-86
120. Park SK, Sanders BG, Kline K. Tocotrienols induce apoptosis in breast cancer cell lines via an endoplasmic reticulum stress-dependent increase in extrinsic death receptor signaling. Breast Cancer Res Treat. 2010;124:361-75
Author contact
![]() Corresponding author: Subhrajit Saha PhD, Assistant Professor, Department of Radiation Oncology, Department of Cancer Biology, The University of Kansas Medical Center, MS 4033, 3901 Rainbow Boulevard, Kansas City, Kansas 66160; Phone: 913-588-1054; Fax 913-588-3663; Email: ssahaedu
Corresponding author: Subhrajit Saha PhD, Assistant Professor, Department of Radiation Oncology, Department of Cancer Biology, The University of Kansas Medical Center, MS 4033, 3901 Rainbow Boulevard, Kansas City, Kansas 66160; Phone: 913-588-1054; Fax 913-588-3663; Email: ssahaedu