Oncomedicine 2018; 3:15-27. doi:10.7150/oncm.22576 This volume Cite
Review
Circulating Tumor Cells in the Parallel Invasion Model Supporting Early Metastasis
Gerhard Hamilton ![]() , Barbara Rath
, Barbara Rath
Society for Research on Biology and Treatment of Cancer (SRBTC), 1160 Vienna, Austria
Received 2017-8-28; Accepted 2017-9-26; Published 2018-2-1
Abstract

Circulating tumor cells (CTCs) seem to comprise metastasis-initiating cells and their count and phenotype represent an indicator of prognosis and response to therapy in cancer patients. In the classical model of tumor dissemination, specialized cells at the invasive front undergo epithelial-mesenchymal transition (EMT) and enter peripheral tumor-coalescing blood vessels to establish metastatic lesions by extravasation of CTCs. Thus, dissemination of cancer cells occurs after prolonged tumor development and following access to neighboring blood vessels via transit through stromal tissue. However, clinical and new experimental data indicate early tumor dissemination immediately after an angiogenic switch has occurred and an intravasation of tumor cells in the tumor core region. Accordingly, CTCs may have direct access through fenestrated and irregular intratumoral vessels and, therefore, the metastasis-initiating cells become active a long time before lesions are detected clinically. This model of cancer cell intravasation is better compatible with the release of CTC clusters and apoptotic CTCs and, furthermore, spares the requirement for an elusive and complicated EMT/MET program. Similarly, the intravasation of large number of CTCs observed in metastatic disease may be alleviated by an intratumoral process instead of a stromal crossing. This early metastasis model changes the possible pathways targeted for the prevention and inhibition of metastasis and the significance of CTCs which are detected and analyzed late after the initial phase of tumor dissemination.
Keywords: Metastasis, Circulating Tumor Cells, Intravasation, Epithelial-Mesenchymal-Transition, Angiogenesis.
Introduction
Most patients with advanced cancer succumb eventually to metastatic disease. [1, 2] Since tumor dissemination is very difficult to study in patients, the individual steps leading to secondary lesions have not been fully characterized. [3] Primary tumors release invasive cells locally which migrate and intravasate into vessels or lymphatics and spread to distant sites in a so-called invasion-metastasis cascade. [4] Thus, metastasis may represent the selective growth of unique subpopulations of malignant cells that preexist within the parent tumor. Different metastases seem to originate from different single cells, indicating a clonal origin. [5] Genetic lineages of metastases can arise early in primary tumors, sometimes long before diagnosis of the primary tumor. [6]
Circulating tumor cells (CTCs) in blood were proposed more than 100 years ago as potential founders of metastatic lesions. [7] CTCs are detectable in most malignancies, particularly at advanced and metastatic stages. Counting and characterization of CTCs has raised great hope for early detection of systemic disease and for individualizing therapy according to the respective patients and tumor characteristics [8]. Research has focused on the sensitive detection of the rare CTCs using approaches depending on cell surface markers or physical properties of the cancer cells. [9] A host of techniques has been described with the CellSearch© system, which selects peripheral cancer cells according to their expression of EpCAM, the single method approved by the FDA so far. [10] CTCs counts were demonstrated to have prognostic significance in several tumor types and an increase under cytotoxic therapy correlates with the resistance to treatment. [11] However, despite a host of techniques developed to count and identify CTCs in the last decade, the clinical applications in diagnosis and therapy are still limited.
Although CTCs were identified and studied in most malignancies, there is still a lack of firm knowledge in regard to their cell biologic characteristics and life cycle. Particularly, the time point of the first release of CTCs, their genetic profile in relation to the bulk tumor, the putative modes of intravasation, their ways of survival in the circulation and possible modes of extravasation are not clear. In most cancer patients, CTCs are rare cells in circulation (a few to a few hundred CTCs per 10 ml blood) and are difficult to detect. Furthermore, CTCs reveal extensive heterogeneity, seem to exhibit poor survival in blood and show an extremely low rate of successful initiation of metastases. These properties have prevented an exhaustive characterization of CTCs in vitro, except in short term cultures and as xenografts in immunocompromised mice [11]. However, small cell lung cancer (SCLC) is distinguished by excessive amounts of CTCs in advanced stage and this exceptional case allowed us to establish 6 permanent CTC lines in vitro which could be employed to study several aspects of the characteristics of CTCs. [12]
Some aspects of CTCs were deduced from a classical model of tumor dissemination which comprises gain of invasive properties of selected clones of tumor cells, intravasation linked to epithelial-mesenchymal transition (EMT), distribution via blood and the lymphatic system and, finally, reversion of EMT via MET and extravasation for colonization at distal sites. [4] Consequently, metastasis would be a relatively late event in tumor development after invasive growth, elicitation of blood vessel extensions and intravasation after a time-consuming crossing of the surrounding tumor stroma. However, clinical observations and recent experimental evidence indicate that CTCs do not arise at the invasive edge of tumors but are released preferentially in the core region. [13] Additionally, this process may occur early in tumor development directly after the angiogenic switch of small primary tumor lesions. This is in line with several clinical observations and hold important consequences for the release and significance of CTCs which are discussed in the present review.
Circulating Tumor Cells (CTCs)
Accurate and repeated biopsies of tumor tissues are crucial for the improved understanding and monitoring of changes in malignant cell populations during disease progression and in response to therapies. Invasive tissue biopsies for monitoring are attempted to be replaced by so-called liquid biopsies which can be easily and frequently repeated and rely on detection of either circulating DNA or cancer cells. Thus, detection and characterization of CTCs poses the potential to estimate the risk for metastatic relapse, to stratify patients to different adjuvant therapy, to identify new therapeutic targets and to monitor systemic anticancer therapies. [8, 14] Direct evidence for a tumorigenic potential of CTCs came from xenograft assays of metastatic breast cancer and small cell lung cancer (SCLC) that is characterized by early dissemination and utterly poor prognosis [15, 16]. In metastatic breast cancer, oligoclonal clusters are held together through plakoglobin-activated adhesion and harbor > 20-fold increased metastatic potential compared to single-cell CTCs [17].
CTCs were described for most tumor types and were demonstrated to have prognostic significance. [18] CTCs are shed from the primary tumor into the circulatory system and act as seeds that initiate cancer metastasis to distant sites. [9] CTCs are rare among blood cells and need to be enriched for further analysis by marker-dependent and -independent methods. The most advanced system for clinical use is the CellSearch© system which relies on immunomagnetic separation of EpCAM-positive tumor cells and confirmation of their cancer origin by proof of expression of cytokeratins and absence of the leucocyte marker CD45. [10, 11] The prognostic threshold for the CellSearch© CTC count for breast, pancreatic and colon cancer amounts to 5, 5 and 3 CTCs/7.5 ml blood, respectively. However, this detection system may miss important EpCAM-negative metastatic CTC subpopulations. Alternative marker-independent methods are based on physical properties of CTCs for discrimination from blood cells such as size, rigidity, surface charge, flow characteristics and others. [19] These techniques result in detection of larger numbers of CTCs compared to the CellSearch© system and the additional cells found may be more aggressive and invasive than EpCAM-positive cancer cells. The multitude of methods to capture CTCs are described in a large number of comprehensive reviews. [9, 20, 21] In addition to CTCs, circulating tumor microemboli (CTM)/CTC clusters, CTM-associated materials (CTMat) and circulating free DNA can be detected in cancer patients. [22] The essential result of the assessment of CTCs in cancer patients is the correlation of higher CTC counts with lower disease-free survival and poorer prognosis as well as a correlation of the decrease of the CTC counts with effective chemotherapy. [11, 20, 23] However, there are a large number of difficulties in detection at different stages of cancer and their phenotypes. It is unclear whether the metastatic cascade results from early dissemination of distinct cells or requires development of fully malignant cells from locally advanced tumors. [24]
In addition to many metastatic patients, CTCs are also reported to be detected in patients with early stage cancers [25-27]. One or more CTCs were reported to be detected in all patients with early-stage prostate cancer, as well as 20% of node-negative and 24% of stage 1-3 breast cancer patients [28-30]. Most results showed that baseline CTC numbers among different cancer types were not correlated with tumor size. A significant variation in the proliferative index (from 1 to 80%) by Ki67 staining of CTCs found in patients at different stages of disease has been described. [31] CTCs are part of disseminated tumor cells (DTCs) and these cell populations are highly correlated, although tests for DTCs in bone marrow were more frequently positive than in blood. [32] This might be explained by the fact that blood analyses allow only a 'snapshot' of tumor cell dissemination. [33] Although cells are shed from a tumor every day, hematogenous metastasis is believed to be very inefficient. In fact, only a small fraction (approximately 0.1%) may remain alive in the circulation after 24h, among which even fewer cells (<< 0.01%) are progenitors of a metastatic mass. This inefficiency is widely supposed to be a result of the destruction of cells in the bloodstream by shear stress and the immune system and a slow rate of extravasation and proliferation in the stroma at a secondary site. [34]
Despite the early detection and possible guidance for the treatment of metastatic spread the widespread clinical usage of CTC tests is limited by the rarity and heterogeneity of CTCs. The major bottleneck here is the rarity of CTCs in the M0 situation and the low blood volume (< 10 mL) usually investigated, insufficient to reliably detect the few CTCs present. [20, 21, 29] Analysis of CTCs is not part of clinical care of tumor patients but a lot of evaluation studies are running in the clinics. That CTCs are undetectable in many metastatic patients and only a minority of CTCs has metastatic potential indicates that CTC enumeration alone is unsatisfactory in disease staging and prognosis. In contrast to cfDNA, CTCs represent intact viable tumor cells that can be analyzed for biological molecules, such as DNA, RNA and proteins. [35] Biomarkers based on the gene expression and genomic profile of CTC subsets that forecast homing and colonization needs to be identified. CTCs and DTCs can be studied in vitro for mechanisms of chemoresistance and in animal models for their metastatic potential. [15, 36, 37] However, to identify the CTCs which have the potential to initiate metastases and to find means to interfere with this process is extremely difficult. From the time of a primary tumor to distant metastasis of 38715 breast cancer patients, a tumor doubling time of 1.7 ± 0.9 months was determined. [38] Fitting the data for of T1B patients yielded an estimate of the metastatic efficiency of 1 metastasis formed per 60 million DTCs. Thus, to reduce the 5-year risk of distant metastasis for TXNXM0 patients from 9.2% to 1.0%, the primary tumor needs to be detected and removed below a diameter of 2.7 ± 1.6 mm. At this size, the model predicts that there will be 9 ± 6 CTC/L blood and, consequently, sensitivity of CTC detection would have to be improved by at least 15-fold and combined with methods that minimizes false positive results.
Patient-derived xenografts (PDX)
A method to expand and to study tumor induction by CTCs is the generation of PDX in immune-compromised mice. For PDX, frequently cells/CTCs derived from metastases are employed, which bypasses the cellular selection processes of metastatic progression and, in fact, models metastasis from metastasis only. [26] For advanced SCLC, CTCs were enriched by removal of leucocytes and the remaining cells were injected with an extracellular matrix preparation subcutaneously into immunocompromised mice. CTCs generated palpable tumors in a time range of 2.4-4.4 months and reflected the patient's response to platinum and etoposide. [16] The numbers of CTCs inoculated correlated with the time to generate palpable tumors and > 400 CTCs per 7.5mL of blood were required for tumor formation in these PDX. Importantly, CTCs and CDXs from individual patients shared genomic alterations, but displayed intratumoral and especially intertumoral heterogeneity. Such heterogeneity is clinically relevant given its impact on treatment, chemoresistance, dissemination and metastases formation in breast cancer and non-small cell lung cancer. [39, 40] For luminal breast cancer, intrafemoral injection of a minimum of > 1,000 human CTCs into immune-compromised NSG mice induced the development of metastases within 6-15 months. However, only 3/106 patients at higher risk of relapses (> 5 CTCs per 7.5mL of blood) actually generated metastases, pointing to difficulties in the extrapolation of numbers from animal models to human disease. [16] Nevertheless, it was suggested that PDX derived from CTC fractions may be suitable for determinations the chemosensivity profile of patients and to help select clinical treatment modalities.
However, xenotransplantation of cell lines that are usually derived from advanced tumors or metastases rely on the correctness of the linear progression model. For the parallel model, high numbers of CTCs indicate an advanced metastatic state and high genetic heterogeneity of the cancer cells. Preservation of the primary chemoresistance in SCLC seems to stem from a special genetically fixed phenotype, as indicated by exome analysis. [41] As a drawback, SCLC PDXs are not applied orthotopically, develop in a foreign murine environment and lack interaction with immune effector cells as well as other human normal tissues. [42] Our own experiment demonstrated tumorigenicity of first two CTC lines (BHGc7 and 10) and high variability of marker expression and chemosensitivity of recultured PDX suspensions in dependence of time to tumor formation and specific localization of the xenografts (unpublished observation). Additionally, the time for development of PDX and testing is too prolonged for a clinical impact for fast-growing tumors and of unproven significance. Data should be obtained from patients rather than animal models because the validity of such animal models to human disease is questionable.
Classical model of CTC intravasation/extravasation
Solid tumor cells could intravasate via both passive and active approaches. [43, 44] However, the frequently used term “shedding” for this release of a subpopulation of tumor cells into the periphery seems to indicate the lack of detailed knowledge of this first step in metastasis. Most CTCs are suggested to be passively pushed by external forces, such as tumor growth and mechanical forces during surgical operation or friction. [45] Very few tumor cells may undergo the epithelial-mesenchymal transition (EMT), such gaining more plasticity and metastatic potential. [46] Current CTC detection methods mostly use the epithelial marker epithelial cell adhesion molecule (EpCAM), which may underestimate CTC number and potentially miss a critical subpopulation. [10] The quantity of CTCs released seems to be correlated to vascularization or invasiveness of the tumor but not to mass.
Frequently, daily shedding of approximately 1-4 million CTCs per gram of tumor tissue is cited but this estimation is based on a single murine model of breast cancer which is unlikely to be transferable to human cancers. [38, 47] While the entry of tumor cells into the circulation is rather common, the process of metastasis is inefficient, seldom exceeding 0.01%. [5] The presence of CTCs does not prove that metastasis has occurred, since CTCs are rapidly eliminated, and trapping in a capillary or the marrow is not predictive of a future colonization. Cellular arrest frequently ends up in apoptosis, dormancy and, rarely, a clinically detectable lesion [5]. Invasive cells may home to bone marrow and survive for prolonged times in a dormant state as DTCs. Metastatic inefficiency is principally determined by failure of solitary cells to initiate growth after extravasation and inability of micrometastases to develop into macroscopic tumors. [48]
That CTCs can be detected in the blood implies that only extremely small and/or deformable CTCs can keep circulating. CTCs clusters are more rapidly cleared from the circulation and have a much higher metastatic potential. [37, 49] Therefore, it is not clear whether actual CTCs are the real source of metastases. CTCs normally should become trapped in capillaries because their size of 20-30 µm is three to four times as large as the diameter of these microvessels. [50] However, the small SCLC CTCs of approximately 8 µm fit into the luminal diameter of capillaries and clusters consisting of several tumor cells were reported to pass capillaries as single-file chain-like structure. [49, 51] Arrest of cancer cells in small capillaries is then followed by the formation of stable attachments, such directing CTCs to specific organs. [52] Liver, or brain whose perfusion is driven mostly by small or defective capillaries are preferred organs for metastatic seeding. [48] Cells that successfully survive and exploit the mechanical permissiveness of the endothelial barrier will eventually seed the distant organ.
EMT
Tumor cells may employ different mechanisms to disseminate in the bloodstream, such as EMT.
EMT designates a developmental regulatory program by which transformed epithelial cells can acquire the capability to invade, to resist cell death, and to disseminate. [43, 53] For single cell and small clusters, migration seems to be much easier after switching to a spindle-cell morphology. Shear stress seems better tolerated by those tumor cells which underwent EMT because tumor cells expressing vimentin and α-actin can adapt to small capillary bores. [48] EMT is suggested to be induced by microenvironmental stimuli distinct at the invasive front, distinct from the factors present in the cores of the tumor. [54] As an inductor, cytokines derived from tumor-associated immune cells seem to activate EMT of cancer cells and their ability to migrate. [55] Tumor-associated macrophages (TAMs) play a critical role in the proliferation, invasion, angiogenesis, and metastasis of a variety of human carcinomas. Tumor cell intravasation occurs in association with perivascular macrophages in mammary tumors in the absence of local angiogenesis. [56] Marker-independent CTC enrichment demonstrate a spectrum of CTC EMT phenotypes with alterations which may be as minor as increases in vimentin and concurrent decreases in E-cadherin without a true cell-type switch. [57] Two other distinct modes of invasion possibly implicated invasion comprise “Collective invasion” and a mode of “amoeboid” invasion. [58] Indeed, some carcinoma cells exhibit a partial mesenchymal state a phenotype absent from normal tissues. [43]
Still, it remains to be determined what fraction of CTCs lose EpCAM expression and undergo (partial) EMT and whether these CTCs have increased metastatic potential or elevated chemoresistance and, therefore, greater prognostic value. The potential relevance of EMT to metastasis is largely based on in vitro studies using transformed epithelial cells and experimental manipulations. [59] Tumors consist of genetically unstable cells and infiltrating cells could contribute to shifts in linage markers. The gain or loss of markers seems insufficient to assume a whole-scale gene expression reprogramming of a cell type.
While the presence of EMT is largely argued based on evidence from in vitro experiments, the in vivo data are unclear. [59] The EMT is primarily considered a phenomenon of the in vitro environment. [60] It is not clear whether alternative regulatory programs can also enable EMT-like capability. Actually, the process by which metastatic cells arise from within populations of non-metastatic cells of the primary tumor is largely unknown. [61] EMT is not often detected in tumor pathological preparations and it remains debatable whether this in vitro model has an in vivo counterpart [59, 62]. That a collection of random gene mutations induce cells which intravasate, evade immune attack, extravasate at distal sites and recapitulate an epithelial phenotype seems unlikely [43, 61]. Moreover, many gene changes associated with EMT can also be found in most non-metastatic benign tumors. [63]
Tumor progression models
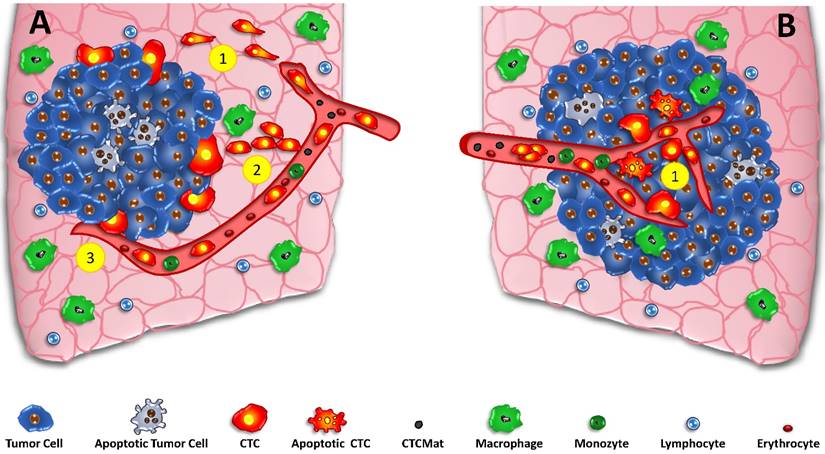
Basically, there are two models for the initiation of the metastatic process, namely the classical linear model and the parallel mode (fig.1) of start of tumor spread. [24] Generation and significance of CTCs detected during metastasis depends critically on the time course and localization of the metastasis. The linear model states that cancer cells pass through multiple successive rounds of mutation and selection for invasiveness within the primary tumor [24, 64]. After a significant time of accumulation of aberrations, these cell clones expand and aggressive cancer cells leave the primary site to seed secondary lesions. CTCs are regarded to develop at the invasive front of the tumors in cooperation with normal cells of the microenvironment (fig.1A). Thus, the metastatic process starts delayed after some time of tumor development and the cells with a putative EMT phenotype must cross the adjacent stroma to intravasate. [65] The assumption that stromal invasion is a prerequisite for spreading suggest the invasive front of the primary tumor as site of intravasation. EMT may constitute a bottleneck for high rates of intravasation but the requirement of this phenotype switch is highly controversial. [59] Such, intravasation is often regarded as a relatively late process during cancer progression, initiated after aggressive cancer cells undergo EMT. [66]
However, the linear cascade model is at odds with amassing evidence indicating that the onset of cancer metastasis occurs much earlier in tumor development than is generally indicated by clinical staging of primary tumors. [67] According to retrospective clinical data, the establishment of clinically relevant metastases can take place at stages preceding substantial local invasion by primary tumors [68]. Furthermore, the parallel progression model accounts for distinct genetic alterations of tumor cells at primary and distal sites due to early separation and independent development. In this line, DTCs can localize in lymph nodes or in the bone marrow prior to the establishment of metastases. The concept of early metastases is also supported by mathematical computation of the time required for distant outgrowths to become life-threatening metastases. [38] The parallel progression model dates back at least to the 1950s, when considerable effort was made to quantify human cancer growth rates (fig.1B). Data suggest that metastasis must be initiated long before the primary tumor was diagnosed, because in respect to their growth rates, metastases were simply too large to be accounted for by initiation at a late stage of primary tumor development. [68, 69]
In detail, tumor volume doubling times (TVDTs) for metastases and primary tumors are comparable, typically approx. 60-200 days but up to two times faster for metastases. [68, 70] In linear progression, dissemination of a metastasis founder cell shortly before surgery would therefore lead to manifestation of a 1 cm metastasis within 6-12 years in breast cancer. Tumor registries report a median time from resection to distant metastasis of 35 and 20 months for patients with T1 (<2 cm) and T3 (>5 cm) tumors, respectively. [71, 72] According to linear progression, growth rates of metastases would have to far exceed that of the primary tumor which is not supported by observations. Parallel progression accounts better for the observed dynamics by setting the beginning of tumor spread to years before diagnosis of a primary tumor. In addition, the parallel progression model predicts DTC clonal diversity in accordance with genetic analysis. Predicting responses to therapies will require the molecular characterization of CTCs/DTCs, which may be significantly different from the primary tumor.
The finding of several large brain metastases and a very small primary tumor, which might even escape the detection by clinical imaging, is common. [73] Furthermore, cancer of unknown primary site accounts for 5-10% of diagnoses in Europe and the United States. [74] This is in line with early migration of carcinoma cells from the initial lesion and onset of metastasis early in the tumor development. The growth of a tumor from initiation to a size of 1 cm which is the limitation of most current imaging tools, requires an average of 12 years and at least 30 doublings from tumor initiation to diagnosis. [4] Murine models also support an early onset of metastasis by demonstrating that distant micrometastases can be initiated from benign tumors, acquiring malignancy at the secondary site independent of primary tumor progression [24, 75] In a mouse model of pancreatic cancer, metastasis-inducing cells were detected in the circulation before malignancy was detected histologically, suggesting that primary tumor cells intravasated ahead of stromal invasion. [25] Importantly, both clinical and experimental studies have provided strong evidence that the angiogenic switch, a prerequisite for intravasation and metastasis, is triggered during the early, pre-invasive stage of tumor development. [76] In conclusion, experimental and clinical data are in favor of the parallel model of the initiation of metastasis which holds important consequences for the underlying mechanisms.
Extravasation
The group of Terstappen l. has developed a mathematical model to estimate the tumor size and the CTC load before the first metastasis has formed from a primary breast cancer tumor. [38] The number of cells disseminated and the efficiency of metastasis formation govern the probability of secondary lesions. The CTC concentration reported for primary breast cancer before surgery is 0.03 CTC/mL (range 0.01-0.05 CTC/mL. [38] The dissemination rate for an 8 mm tumor is 280 CTC/h · g tumor (range 90-470 CTC/h · g tumor) yielding a metastatic efficiency of approximately 60 million disseminated cells per macrometastasis formed. The dissemination rate measured in the efferent vein of human colorectal and renal cancer gave a median estimate of 3,100 CTC/h · g tumor (range of 90-78,000 CTC/h · g). Although metastatic efficiency has not yet been determined in humans a comparable metastatic rate of 0.011%, primarily caused by the failure to establish a macrometastasis, seems to apply. This is substantially less efficient than the murine model median estimate of 1 metastasis in 14,000 DTCs (range 1 in 170 to 1 in 1 million). The large difference of metastatic efficiency between murine model and humans may be attributed to the use of highly metastatic cell lines, difference in size and the immunodeficiency of mouse models. Survival of extravasated cells beyond 2 weeks is estimated between 4% and 50% in the murine model and these dormant cells scattered throughout the body may be reactivated at a later time.
Early metastasis and CTCs
Intravasation can be initiated early and proceed in parallel to or independent of tumor invasion into surrounding stroma, suggesting onset of cancer metastasis much earlier in tumor development than is indicated by conventional clinical staging of primary tumors. [13, 67]. For example, in a murine model pancreatic cancer cells intravasated and colonized the liver even before any primary tumor was detectable, indicating CTCs are present in early-stage cancers. [25] In line, single DTCs can be found in the lymph nodes or bone marrow of healthy women with a history of early-stage breast cancer that have no clinical evidence of metastasis or tumor recurrence. [13]
Tumor cells may enter the circulation as a result of passive shedding or by an active mechanism called EMT. [7, 43] Passive shedding is supposed to occur from the early stages of tumor formation and a large number of tumor cells may disseminate into the blood circulation in this way. [77] Peaks of CTCs shedding seem to correspond to specific events in tumor development, such as the angiogenic switch which is triggered during the early, pre-invasive stage of tumor development. [76] Intravasation of invasive cancer cells is facilitated by the leakage of blood vessels occurring during the tumor neovascularization process. [78] Invasive cancer cells could also enter the blood stream by direct transcellular intravasation through squeezing between endothelial cells and pericytes. Cytoskeletal and membrane remodeling in vessels create a pore-like structure for cancer cells crossing the endothelial cell barrier. [52] Recently, a novel mechanism of extravasation, programmed necrosis (necroptosis) of endothelial cell induced by tumor cells was described. [79] Likewise, the generation of CTC clusters is believed to result from collective migration of tumor cells and intravasation via a leaky vessel in the primary tumor.
Intravasation is a complex process that cannot be fully reproduced in vitro and is rarely observed in vivo. [56] Therefore, intravasation levels are determined by indirect methods such as quantifying vascular-arrested tumor cells in distal tissues or CTCs in the peripheral blood. Intravital imaging of primary tumors in experimental animals did offer access to tumor cell intravasation but, so far, intravasation events could not be monitored across the entire tumors. Deryugina and Kiosses investigated the localization of intravasation within primary tumors in both, a mouse ear and avian chorioallantoic membrane mesoderm live model employing fibrosarcoma and carcinoma cells. [13] They demonstrated that intravasation takes place almost exclusively within the tumor core and involves intratumoral vasculature. [13, 80, 81] In detail, GFP-tagged human fibrosarcoma cell variants were inoculated into the chorioallantoic membrane mesoderm of chick embryos and the vast majority of intravasated cells localized to the core of primary tumors. [13]. The bulk of intravasation events was found within the new immature blood vessels in the core of the primary tumor, uncoupled from intravasation localized to the invasive front. Clearly, vasculotropic cancer cells which invade tumor-adjacent stroma and migrate to tumor-coalescing vessels play a minor part in the cancer spread. As a consequence, intratumoral localization of intravasation forwards the generation of metastases during cancer progression in patients much earlier than appreciated previously, immediately after the first angiogenic vessels are formed.
Newly formed intratumoral vessels transport oxygen and nutrients to the tumor cells. These blood vessels are typically aberrant as tumor neovasculature is marked by convoluted vessel branching, distorted and enlarged vessels, intermittent flow, fenestration, leakiness, and abnormal levels of endothelial cell proliferation and apoptosis. [78] The cut-off size of the putative pores in the walls of tumor vessels varied from ~100 nm to 2 µm depending on the tumor type and whether it is growing or regressing. [45, 82] Permeability of the vessel walls facilitates the intravasation of cancer cells. [58] In combination, vessel permeability and hypoxia data indicate that tumor cell spread occurs within the primary tumor core via leaky, permeable vessels, which provide enough oxygenation to sustain intravasation ability of tumor cells without aid of hypoxia-induced gene expression. Angiogenesis is induced surprisingly early during the development of invasive cancers in animal models and in humans. Early triggering of the angiogenic switch has been found by analyses of premalignant lesions, including dysplasias and in situ carcinomas of different organs. [83] The peritumoral inflammatory cells help to trip the angiogenic switch in previously quiescent tissue and to sustain ongoing angiogenesis associated with tumor growth.
Early dissemination and CTCs
Detection of early metastatic disease
CTCs are rare in non-metastatic patients and, therefore, detection of dissemination at an early stage is difficult, especially using smaller amounts of blood. [23] Whereas metastasis via the lymphatic route usually takes longer until distant metastases are set, spreading via blood vessels rapidly induces distant metastases. [73] Although it is stated that tumors release millions of cells per day, this estimation is based on a single experimental animal model which is no realistic representation of a human lesion. [47] Furthermore, the threshold of a poorer prognosis for colon, breast and prostate cancer patients with 3-5 CTCs/7.5 ml blood by the CellSearch© system point to a limited shedding of cells from such tumors. [10] The kinetics of the release of tumor cells into the circulation is unclear and at certain time points there will be no CTCs at all or they will be missed by sampling a blood sample of insufficient size. In case of COPD, detection of premalignant cells which developed into cancer several years later was reported for a single case and a respective clinical trial is ongoing. [84] However, the chance of a malignant disease in COPD patients is approximately 1 in 200 patients and such a high number of possibly affected people will have to be screened with considerable cost and effort to find a few patients at high risk. [85] The number of false-negative patients is expected to be rather high due to the scarcity of the CTCs and technical difficulties in their detection. In the parallel model of metastasis, small tumors and low tumor cell spreading will make the detection of CTCs very demanding.
Identification of metastasis-inducing cells
CTCs are the founding cells of metastases and since metastatic disease is the main cause of cancer mortality prevention of tumor dissemination is regarded as essential contribution to make malignancies curable. One of the goals of CTC analysis is the identification of the metastasis-inducing cells. This search has to take place during the disease course in advanced and metastatic cancer patients when invasive cancer cells are still circulating according to the linear tumor dissemination model. However, as a consequence of early dissemination, the original metastasizing CTCs are no longer existent at a time point of the clinical detection of disease and the CTCs may have a different phenotype. In accordance with the parallel metastasis model, the first CTCs are released at a small tumor size following the first angiogenic switch. [13] Oxygen can diffuse over a distance of approximately 200 µm and beyond that distance vessels are needed for respiration [86]. Thus, it may be not possible in patients to identify the first step of metastasis and isolate the respective cancer cells. Furthermore, it may be not feasible to prevent the first step of metastasis by interference with specific signals and mechanisms of CTCs. At later phases the CTCs released by the clinically detected lesions are supposed to have further developed and genetically acquired a different phenotype compared to the initiation of metastasis and may stem from DTCs or released by secondary lesions.
Role of EMT in the parallel model of metastasis
Release of the CTCs in the tumor core according to the parallel metastasis model may provide an easy access to tumor vessels without the need to pass a significant distance through the tumor stroma, as in the case of shedding from the invasive front. [43] Since the newly built tumor vessels are irregular and fenestrated they may offer a facilitated access to the circulation by mechanisms of transepithelial migration, necropsis, physical pressure or proteolytic invasion. [77, 87] There may be no need to switch to a motile mesenchymal phenotype or to rely on collective or amoeboid migration. This enhanced and direct intravasation may explain, why, despite the shedding of millions of CTCs, cancer cells with an EMT phenotype adjacent to the tumor were rarely found in patients. Thus, EMT and its reversal at metastasis target sites, MET, seem not to be necessary for tumor spread. [61] Furthermore, our results with SCLC CTCs have shown that these cells themselves produce angiogenic factors, like CHI3L1 and VEGF, as well as proteolytic enzymes (MMPs, cathepsins) and have the means to elicit angiogenesis and intravasate on their own. [88]
Origin of CTCs from a partially hypoxic tumor core may account for the release of cells with variable viability and proliferative state. Part of the cells will not perish in the circulation but may be apoptotic in the beginning. Release of CTCs from the tumor core fits better to the occurrence of non-proliferating or apoptotic tumor cells since at the place of their origin intermittent hypoxia is expected to prevent normal cell growth. Furthermore, intravasation in the tumor core is expected to be facilitated for group of cells in contrast to a complicated migration through stromal tissue. Additionally, part of the CTCmat may be released directly from the core of the tumor and not in reaction to the adverse conditions in the circulation. In contrast, CTCs which are supposed to actively cross tumor stroma should be fully viable and most likely in a proliferative phase of the cell cycle. SCLC occurs in most of the cases in smokers with high tobacco consumption and the cancer cells are riddled with thousands of mutations. [89] In this situation, the precisely timed acquisition of a mesenchymal cell type and its controlled reversal during extravasation is an unlikely event. [61] Furthermore, reduction in cell adhesion-associated proteins and an increase in vimentin, the so-called partial EMT, is not indicative of a real cell type switch.
Extravasation in the parallel model of metastasis
In order to establish secondary lesions, CTCs need to extravasate, colonize distinct sites and grow to full metastases. For the linear tumor model, tumor cells which exhibit an EMT phenotype will have to switch back to the epithelial type again in a process called MET since metastases are of epithelial phenotypes again (fig.1A). Whether a dysregulated cancer cell with a high mutation burden is capable to execute a precise forward and reverse phenotypic switch is under discussion. [61] In an experimental lung cancer model, EMT was found not to be necessary for metastasis but related eventually to drug resistance. [90] The low metastatic efficiency of CTCs was found to be associated with the low frequency for malignant growth in the invaded organs.
In the parallel mode, dissemination occurs at a small size of the primary tumor and seems not to require EMT for intravasation (fig.1B). Clearly, EMT is not necessary for motility in the circulation but it has been claimed that this phenotype enhances the resistance of epithelial cells to shear stress. [91] As a consequence of early dissemination, cancer cells are shed for a longer time period and the chance to accumulate the threshold of metastasis-initiating events is increased. [92] According to observations in breast cancer patients approximately 60 million CTCs are needed for the induction of one metastasis. [36] In case of SCLC CTCs, smaller cell clusters may become stuck in capillaries and invade through exertion of physical pressure and with the help of their own proteases produced. For SCLC CTCs, the preferred capillaries are situated in liver and brain. [93] Although most CTCs are supposed to perish in the circulation, Naumov et al. have quantified breast cancer cell fate using an intravital microscopy and found that the majority of cells not only survived, but extravasated and persisted as single dormant tumor cells. [94] Wong et al. used intravital microscopy to demonstrate that, in their breast cancer metastasis model, the majority of cells not only remained intravascular but began to proliferate intralumenally. [95] These experiments showed that metastatic cells derived by spontaneous metastases were intravascular, and that early colony formation was intravascular. This model for pulmonary metastasis in mice demonstrates that tumor cells can attach to lung endothelium soon after arrival, proliferate intravascularly and extravasation of the tumor occurs by outhgrowth of intravascular micrometastatic foci.
Genetic and phenotypic makeup of CTCs
Clearly, the CTCs found in metastatic patients differ from the bulk of primary tumors and possibly from metastases themselve and such may represent a poor surrogate marker for response to therapy. [92] For example, the SCLC CTCs proved to be chemosensitive but the tumors which have relapsed exhibit a broad drug resistance. [12, 96] Therefore, successful reduction or elimination of the CTCs is not certified to correlate with response of the resident lesions. In 65% of cases of colorectal cancer, lymphatic and distant metastases were derived from independent subclones in the primary tumor, whereas in 35% of cases they shared a common origin. [97] Single cell genetic and transcriptomic analysis of CTC is now feasible to assess the heterogeneity of these cancer cells although detailed analysis is difficult due to possible faults introduced through amplification of the minute amounts of DNA/RNA samples. Since calculations show that a metastasis is formed by less than 0.01% of the CTCs and after circulation of approximately 60 million CTCs in breast cancer, single-cell analysis to identify the true metastasis-inducing subpopulation seems to be demanding. In addition, processes directing cell adhesion and interaction may be challenging to be identified at the level of the transcriptome. One solution may be the in vitro expansion of viable CTCs and there genetic and phenotypic characterization. [12]
Surrogate marker of response
Systemic anticancer therapy targets tumor cells that have been shed from the primary lesion to settle elsewhere and which are undetectable by clinical imaging and inaccessible to surgery. [24] The use of primary tumors to predict therapy response is based largely on the linear progression model in which tumor ontogeny proceeds within the original tumor microenvironment and, consequently, the primary tumor determines the molecular characteristics of DTCs. In parallel progression, tumor cells depart the primary lesion before the acquisition of fully malignant phenotypes to undergo somatic development and metastatic growth at a distant site. [92] The early dissemination and deviating genomic evolution of DTCs versus primary tumors in metastasis such challenges the importance of the primary tumor for prediction of the response to therapy. The assumption that CTCs are valid surrogate marker of the bulk tumors seems to be questionable. Although the primary tumor in SCLC is largely chemosensitive and the CTCs are responsive, in relapsed patients the tumor is broadly chemoresistant. [12] Although, persistent high CTC counts in response to chemotherapeutic treatments seem to indicate drug resistance of the resident lesions, a drop of the CTC count may still not be representative for the tumor as whole. Furthermore, detection of drug resistance via CTC count in clinical decision making is without effect in the unavailability of alternative effective treatment regimens. [98]
SCLC
To study the cell biology of CTCs in detail, cell cultures of these cells need to be established and SCLC has offered the best chance due to the high numbers of CTC present and their marked metastatic activity. SCLC is an aggressive neuroendocrine tumor which is disseminated in most cases at first presentation and has a 2-year survival rate below 10%. [99] Despite high response rates to first-line chemotherapy consisting of platinum drugs/etoposide combinations, the tumor usually recurs within approximately one year in a chemoradioresistant form. In contrast to most other tumors, SCLC exhibits large numbers of CTCs with a mean value of 400 CTCs/7.5 ml blood and with extreme values of up to several thousand cells/7.5 ml blood in individual patients. [16] High blood circulation, aggressive growth and inflammation linked to recruitment of macrophages seem to contribute to tumor cell shedding. [100] The SCLC CTCs are comparable small (approximately 8 µm) and, thus, can more easily pass capillaries and recirculate. These unique characteristics allowed us to set up a panel of 8 permanent SCLC CTC line from blood samples which could be used to study their cell biologic traits. So far, a few CTC cell lines have been established from breast and one colon cancer patients. The cancer stem cell hypothesis assumes that rare chemoresistant stem cells survive the initial chemotherapy and are eventually responsible for tumor relapses.[101] However, SCLC CTCs lack stem cell markers, such as CD133, CD44, ABCG2 and exhibit a shift towards an epithelial-mesenchymal phenotype without a real cell-type switch. [57, 102] Moreover, the SCLC CTCs are chemosensitive, except one case with primary platinum resistance, and form large multicellular aggregates, termed tumorospheres, which contain quiescent and hypoxic cells. These 3D-structures are highly chemoresistant compared to the same cells as single cell suspensions. [12] Although the CTC lines can be kept in culture for years, formation of apoptotic fragments is observed, similar to CTCmat detectable in the circulation. In conclusion, SCLC CTCs circulate in high numbers, lack EMT and CTC phenotypes and have high metastatic potential. All CTC cell lines express proteases, VEGF and the angiogenic inductor CHI3L1 which enables the cells to induce neoangiogenesis and to intravasate. [88]
CTCs and the linear and parallel models of metastasis. In the classical parallel metastasis model (A) the tumor grows and special cells of the invasive front detach, undergo EMT and intravasate into tumor-coalescing blood vessels (A1) Alternative crossing of the stroma may be done by collective migration (A2) and direct intravasation by necropsis of endothelia (A3). Contrary, the parallel model of metastasis incorporates early dissemination in the core of the developing tumor (B1). CTCs and possibly apoptotic tumor cells and CTCmat are released directly into the irregular vessels of small tumors without the need to undergo EMT and to travel through tumor stroma at the invasive front to gain access to vessels.
Discussion
Liquid biopsies in form of CTC analysis hold great potential for detection and proof of malignant disease, monitoring of the course of the malignancy and evaluation of the response to treatment for guidance of anticancer therapy. [23] Whereas the development of techniques for enrichment and identification of CTCs are progressing fast, characterization of their biologic significance and their precise role in cancer dissemination is lagging behind. Research on CTCs is hindered by the rarity and heterogeneity of these cancer cells and the infrequence of short-or long-term cultured specimen. Importance and specific functions of the CTCs at distinct time points in tumor spread are related to the details of the metastatic process. The significance of CTCs in tumor dissemination depends on the validness of the linear or parallel scheme of metastasis. [12] Clinical observations, calculations of tumor growth velocities and new experimental models are in favor of the parallel progression model, pointing to early tumor spread upon the first angiogenic switch in small tumors, prior to the deferred detection by clinical imaging methods. As further consequence, in this model intravasation occurs preferentially in the tumor core, facilitating direct entering of intratumoral vessels by cancer cells. [13] In such a model, CTCs appear early, long before the tumor or metastases are detected in patients by symptoms, biomarkers and imaging. Furthermore, dissemination, extravasation and colonization of distal organs may have happened already at the time of first presentation in clinics. Consequently, the truly metastasis-inducing CTCs may be not observable and their phenotype no longer accessible. Because metastasis has occurred in many patients at first presentation, therapeutic modalities which target the tumor cell invasion and other early processes of metastasis may not have an impact on final outcomes. [4] Therefore, for this large group of patient the prevention of metastatic colonization, the progress from micrometastatic tumor cells to overt metastases, seems to constitute a promising target. [103]
In respect to the “shedding” of cancer cells into the circulation, switching to a mesenchymal phenotype (EMT) seems not to be necessary in the parallel model of metastasis with release of the cells from tumor cores at small tumor sizes. This supports the intravasation of large number of tumor cells, the occurrence of cell clusters and the possible discharge of apoptotic and fragmented cells into irregular and fenestrated tumor vessels. A large number of CTCs are found apoptotic and the higher tumorigenic potential of CTC clusters may in part be due to their functional intactness. For the SCLC CTC clusters, we found marked attachment to clots from platelet-rich plasma (unpublished observation) and, accordingly, clotting plays an important role in cancer progression and metastasis. [104] The inversion of EMT, namely MET, for extravasation is unnecessary in the parallel tumor model and this spares the execution of complicated phenotype switches in cancer cells carrying a high mutational load. [61]
In a parallel metastasis model, some cells depart early from the primary and develop into genetically diverse CTCs and metastases. The precise phenotype of a CTC is influenced by specific microenvironmental conditions in interaction with normal cell types such as stromal fibroblasts, inflammatory immune cells, blood cells, bone marrow cells and others. [100] Therefore, the assumption that CTCs are representative of the bulk tumors is questionable. For example, in metastatic SCLC the CTCs are chemosensitive but responses of the bulk tumor to chemotherapy are poor leading ultimately to a dismal prognosis. In this case, CTCs are able to form aggregates, designated tumorospheres, which exhibit a broad chemoresistance. [12] Of course, CTCs are easily accessible to high concentrations of chemotherapeutics in the circulation, in contrast to poorly vascularized tumors holding a specific microenvironment which contributes to chemoresistance. Therefore, a drop in CTC counts in response to treatment must not necessarily translate to a tumor response and serve as a valid surrogate marker.
Generation of PDX and metastases in immunocompromised mice is short of a full proof of a metastasis-inducing capacity in humans. The influence of microenvironmental conditions limits the value of PDX models. CTCs are applied subcutaneously and grow in a murine milieu of growth factors and conditions in the absence of interaction with immune effector cells. Although the cancer cells retain some basic markers and features, which subpopulation of the heterogeneous CTCs expands under this setting is not clear and may not be directly related to the metastasis-inducing cells in humans. Furthermore, generation of xenografts seem to require a CTC count > 400 CTCs/7.5 ml blood in patients and can thus be done in advanced metastasis only and, for example, no new and effective therapy was inferred from PDX models in SCLC so far.
In conclusion, a large number of methods have been devised to detect, enrich and characterize CTCs but the FDA-approved CellSearch© may fail some important cells and the other methods are not standardized. Numerous studies in different tumor types have demonstrated a negative impact on higher CTC counts on disease-free and overall-survival in cancer patients and resistance to therapy in patients which fail to show a drop in CTC counts in response to treatment. However, CTCs are mostly detectable late in metastasis in advanced disease and their heterogeneity complicates the correlation of counts and phenotypes as well as cell biologic characteristics and relevant markers. For example, in almost half of patients with progressive metastatic breast cancer CTCs could not be detected using the CellSearch system. [15] The current strategies to detect rare CTCs from blood samples of limited size at early stages of tumor development are not sensitive enough and normal epithelial cell types, especially in inflammation, make this task more difficult and will give false positive results. Sensitivity of the detection of CTCs can be increased by diagnostic leukapheresis but at increased expenditure. [105, 106]
In comparison to classical tumor markers, the monitoring of CTCs is technically demanding and has yet to prove its value in clinical decision making and patient care. Availability of a larger panel of CTC lines, as in SCLC, will help to investigate the role of CTCs in disease progression and chemoradioresistance in metastatic patients. [12] Studies on CTCs will have to move further from assessment of counts to relevant markers associated with resistance, intravasation and interaction with the specific microenvironments encountered. [107] Analysis of CTCs can help to identify patients a high risk, to differentiate benign from malign disease and guide therapy; however, the limitations and intricacies of the interpretations of the cancer cells detected has to be considered comprehensively. [108]
Competing Interests
The authors have declared that no competing interest exists.
References
1. Eccles SA, Welch DR. Metastasis: recent discoveries and novel treatment strategies. Lancet. 2007;369:1742-57
2. Wan L. et al. Tumor metastasis: moving new biological insights into the clinic. Nature medicine. 2013;19:1450-64
3. Brabletz T. et al. Roadblocks to translational advances on metastasis research. Nature medicine. 2013;19:1104-9
4. Talmadge JE, Fidler IJ. AACR centennial series: the biology of cancer metastasis: historical perspective. Cancer research. 2010;70:5649-69
5. Fidler IJ. Cancer biology is the foundation for therapy. Cancer Biol Ther. 2005;4:1036-9
6. Zhao ZM, Zhao B, Bai Y. et al. Early and multiple origins of metastatic lineages within primary tumors. Proc Natl Acad Sci U S A. 2016;113:2140-5
7. Caixeiro NJ, Kienzle N, Lim SH. et al. Circulating tumour cells-a bona fide cause of metastatic cancer. Cancer Metastasis Rev. 2014;33:747-56
8. Chen L, Bode AM, Dong Z. Circulating Tumor Cells: Moving Biological Insights into Detection. Theranostics. 2017;7:2606-19
9. Rawal S, Yang YP, Cote R, Agarwal A. Identification and quantitation of circulating tumor cells. Annual Review of Analytical Chemistry. 2017;10:321-43
10. Andree KC, van Dalum G, Terstappen LW. Challenges in circulating tumor cell detection by the CellSearch system. Mol Oncol. 2016;10:395-407
11. Bardelli A, Pantel K. Liquid Biopsies, What We Do Not Know (Yet). Cancer Cell. 2017;31:172-9
12. Klameth L, Rath B, Hochmaier M. et al. Small cell lung cancer. model of circulating tumor cell tumorospheres in chemoresistance. Sci Rep. 2017;7:5337
13. Deryugina EI, Kiosses WB. Intratumoral Cancer Cell Intravasation Can Occur Independent of Invasion into the Adjacent Stroma. Cell Rep. 2017;19:601-16
14. Kaiser J. Medicine. Cancer's circulation problem. Science. 2010;327:1072-4
15. Baccelli I, Schneeweiss A, Riethdorf S. et al. Identification of a population of blood circulating tumor cells from breast cancer patients that initiates metastasis in a xenograft assay. Nat Biotechnol. 2013;31:539-44
16. Hodgkinson CL, Morrow CJ, Li Y. et al. Tumorigenicity and genetic profiling of circulating tumor cells in small-cell lung cancer. Nat Med. 2014;20:897-903
17. Aceto N, Bardia A, Miyamoto DT. et al. Circulating tumor cell clusters are oligoclonal precursors of breast cancer metastasis. Cell. 2014;158:1110-22
18. Wang J, Chang S, Li G, Sun Y. Application of liquid biopsy in precision medicine: opportunities and challenges. Front Med. 2017
19. Gabriel MT, Calleja LR, Chalopin A. et al. Circulating Tumor Cells: A Review of Non-EpCAM-Based Approaches for Cell Enrichment and Isolation. Clin Chem. 2016;62:571-81
20. Joosse SA, Gorges TM, Pantel K. Biology, detection, and clinical implications of circulating tumor cells. EMBO Mol Med. 2015;7:1-11
21. Alix-Panabières C, Pantel K. Challenges in circulating tumour cell research. Nat Rev Cancer. 2014;14:623-31
22. Hou JM, Krebs MG, Lancashire L. et al. Clinical significance and molecular characteristics of circulating tumor cells and circulating tumor microemboli in patients with small-cell lung cancer. J Clin Oncol. 2012;30:525-32
23. Rolfo C, Castiglia M, Hong D. et al. Liquid biopsies in lung cancer: the new ambrosia of researchers. Biochim Biophys Acta. 2014;1846:539-46
24. Klein CA. Parallel progression of primary tumours and metastases. Nat Rev Cancer. 2009;9:302-12
25. Rhim AD, Mirek ET, Aiello NM. et al. EMT and dissemination precede pancreatic tumor formation. Cell. 2012;148:349-61
26. Hüsemann Y, Klein CA. The analysis of metastasis in transgenic mouse models. Transgenic Res. 2009;18:1-5
27. Eyles J, Puaux AL, Wang X. et al. Tumor cells disseminate early, but immunosurveillance limits metastatic outgrowth, in a mouse model of melanoma. J Clin Invest. 2010;120:2030-9
28. Nagrath S, Sequist LV, Maheswaran S. et al. Isolation of rare circulating tumour cells in cancer patients by microchip technology. Nature. 2007;450:1235-9
29. Rack B, Schindlbeck C, Juckstock J. et al. Circulating tumor cells predict survival in early average-to-high risk breast cancer patients. J Natl Cancer Inst. 2014:106
30. Lucci A, Hall CS, Lodhi AK. et al. Circulating tumour cells in non-metastatic breast cancer: a prospective study. Lancet Oncol. 2012;13:688-95
31. Stott SL, Lee RJ, Nagrath S. et al. Isolation and characterization of circulating tumor cells from patients with localized and metastatic prostate cancer. Sci Transl Med. 2010;2:25ra3
32. Muller V, Stahmann N, Riethdorf S. et al. Circulating tumor cells in breast cancer: correlation to bone marrow micrometastases, heterogeneous response to systemic therapy and low proliferative activity. Clin Cancer Res. 2005;11:3678-85
33. Xu L, Shamash J, Lu YJ. Circulating Tumor Cells: A Window to Understand Cancer Metastasis, Monitor and Fight Against Cancers. Journal of Cancer Research Updates. 2015;4:13-29
34. Leblanc R, Peyruchaud O. Metastasis: new functional implications of platelets and megakaryocytes. Blood. 2016;128:24-31
35. Heitzer E, Auer M, Ulz P. et al. Circulating tumor cells and DNA as liquid biopsies. Genome Med. 2013;5:73
36. Khoo BL, Grenci G, Jing T. et al. Liquid biopsy and therapeutic response: Circulating tumor cell cultures for evaluation of anticancer treatment. Sci Adv. 2016;2:e1600274
37. Yu M, Bardia A, Aceto N. et al. Cancer therapy. Ex vivo culture of circulating breast tumor cells for individualized testing of drug susceptibility. Science. 2014;345:216-20
38. Coumans FA, Siesling S, Terstappen LW. Detection of cancer before distant metastasis. BMC Cancer. 2013;13:283
39. Kimbung S, Loman N, Hedenfalk I. Clinical and molecular complexity of breast cancer metastases. Semin Cancer Biol. 2015;35:85-95
40. Morrow CJ, Trapani F, Metcalf RL. et al. Tumourigenic non-small-cell lung cancer mesenchymal circulating tumour cells: a clinical case study. Ann Oncol. 2016;27:1155-60
41. Carter L, Rothwell DG, Mesquita B. et al. Molecular analysis of circulating tumor cells identifies distinct copy-number profiles in patients with chemosensitive and chemorefractory small-cell lung cancer. Nat Med. 2017;23:114-9
42. Park D, Wang D, Chen G, Deng X. Establishment of Patient-derived Xenografts in Mice. Bio Protoc. 2016;6:e2008
43. Nieto MA, Huang RY, Jackson RA, Thiery JP. EMT: 2016. Cell. 2016;166:21-45
44. Tsuji T, Ibaragi S, Hu GF. Epithelial-mesenchymal transition and cell cooperativity in metastasis. Cancer Res. 2009;69:7135-9
45. McDonald DM, Baluk P. Significance of blood vessel leakiness in cancer. Cancer Res. 2002;62:5381-5
46. Barriere G, Fici P, Gallerani G. et al. Circulating tumor cells and epithelial, mesenchymal and stemness markers: characterization of cell subpopulations. Ann Transl Med. 2014;2:109
47. Butler TP, Gullino PM. Quantitation of cell shedding into efferent blood of mammary adenocarcinoma. Cancer Res. 1975;35:512-6
48. Luzzi KJ, MacDonald IC, Schmidt EE. et al. Multistep nature of metastatic inefficiency: dormancy of solitary cells after successful extravasation and limited survival of early micrometastases. Am J Pathol. 1998;153:865-73
49. Au SH, Storey BD, Moore JC. et al. Clusters of circulating tumor cells traverse capillary-sized vessels. Proc Natl Acad Sci U S A. 2016;113:4947-52
50. Chaffer CL, San Juan BP, Lim E, Weinberg RA. EMT, cell plasticity and metastasis. Cancer Metastasis Rev. 2016;35:645-654
51. Langley RR, Fidler IJ. The seed and soil hypothesis revisited-the role of tumor-stroma interactions in metastasis to different organs. Int J Cancer. 2011;128:2527-35
52. Reymond N, d'Água BB, Ridley AJ. Crossing the endothelial barrier during metastasis. Nat Rev Cancer. 2013;13:858-70
53. Polyak K, Weinberg RA. Transitions between epithelial and mesenchymal states: acquisition of malignant and stem cell traits. Nat Rev Cancer. 2009;9:265-73
54. Huang D, Du X. Crosstalk between tumor cells and microenvironment via Wnt pathway in colorectal cancer dissemination. World J Gastroenterol. 2008;14:1823-7
55. Suarez-Carmona M, Lesage J, Cataldo D, Gilles C. EMT and inflammation: inseparable actors of cancer progression. Mol Oncol. 2017;11:805-23
56. Wyckoff JB, Wang Y, Lin EY. et al. Direct visualization of macrophage-assisted tumor cell intravasation in mammary tumors. Cancer Res. 2007;67:2649-56
57. Hamilton G, Hochmair M, Rath B. et al. Small cell lung cancer: Circulating tumor cells of extended stage patients express a mesenchymal-epithelial transition phenotype. Cell Adh Migr. 2016;10:360-7
58. Friedl P, Wolf K. Plasticity of cell migration: a multiscale tuning model. J Cell Biol. 2010;188:11-9
59. Jolly MK, Ware KE, Gilja S. et al. EMT and MET: necessary or permissive for metastasis? Mol Oncol. 2017;11:755-69
60. Bacac M, Stamenkovic I. Metastatic cancer cell. Annu Rev Pathol. 2008;3:221-47
61. Seyfried TN, Huysentruyt LC. On the Origin of Cancer Metastasis. Crit Rev Oncog. 2013;18:43-73
62. Garber K. Epithelial-to-mesenchymal transition is important to metastasis, but questions remain. J Natl Cancer Inst. 2008;100:232-3
63. Larue L, Bellacosa A. Epithelial-mesenchymal transition in development and cancer: role of phosphatidylinositol 3′ kinase/AKT pathways. Oncogene. 2005;24:7443-54
64. Cairns J. Mutation selection and the natural history of cancer. Nature. 1975;255:197-200
65. Kalluri R, Weinberg RA. The basics of epithelial-mesenchymal transition. J Clin Invest. 2009;119:1420-8
66. Valastyan S, Weinberg RA. Tumor metastasis: molecular insights and evolving paradigms. Cell. 2011;147:275-92
67. Turajlic S, Swanton C. Metastasis as an evolutionary process. Science. 2016;352:169-75
68. Friberg S, Mattson S. On the growth rates of human malignant tumors: implications for medical decision making. J Surg Oncol. 1997;65:284-97
69. Collins VP, Loeffler RK, Tivey H. Observations on growth rates of human tumors. Am J Roentgenol Radium Ther Nucl Med. 1956;76:988-1000
70. Spratt JS, Meyer JS, Spratt JA. Rates of growth of human neoplasms. J Surg Oncol. 1996;61:68-83
71. Engel J. et al. The process of metastasisation for breast cancer. Eur J Cancer. 2003;39:1794-806
72. Jones T, Neboori H, Wu H. et al. Are breast cancer subtypes prognostic for nodal involvement and associated with clinicopathologic features at presentation in early-stage breast cancer? Ann Surg Oncol. 2013;20:2866-72
73. Popper HH. Progression and metastasis of lung cancer. Cancer Metastasis Rev. 2016;35:75-91
74. van de Wouw AJ, Janssen-Heijnen ML, Coebergh JW, Hillen HF. Epidemiology of unknown primary tumours; incidence and population-based survival of 1285 patients in Southeast Netherlands, 1984-1992. Eur J Cancer. 2002;38:409-13
75. Podsypanina K, Du YC, Jechlinger M. et al. Seeding and propagation of untransformed mouse mammary cells in the lung. Science. 2008;321:1841-4
76. Folkman J. Role of angiogenesis in tumor growth and metastasis. Semin Oncol. 2002;29:15-8
77. Bockhorn M, Jain RK, Munn LL. Active versus passive mechanisms in metastasis: do cancer cells crawl into vessels, or are they pushed? Lancet Oncol. 2007;8:444-8
78. Carmeliet P, Jain RK. Principles and mechanisms of vessel normalization for cancer and other angiogenic diseases. Nat Rev Drug Discov. 2011;10:417-27
79. Strilic B, Yang L, Albarrán-Juárez J. et al. Tumour-cell-induced endothelial cell necroptosis via death receptor 6 promotes metastasis. Nature. 2016;536:215-8
80. Deryugina EI, Quigley JP. Tumor angiogenesis: MMP-mediated induction of intravasation- and metastasis-sustaining neovasculature. Matrix Biol. 2015;44-46:94-112
81. Bobek V, Kolostova K, Pinterova D. et al. A clinically relevant, syngeneic model of spontaneous, highly metastatic B16 mouse melanoma. Anticancer Res. 2010;30:4799-803
82. Jain RK, Munn LL, Fukumura D. Measuring vascular permeability in mice. Cold Spring Harb Protoc. 2013;2013:444-6
83. Raica M, Cimpean AM, Ribatti D. Angiogenesis in pre-malignant conditions. Eur J Cancer. 2009;45:1924-34
84. Ilie M, Hofman V, Long-Mira E. et al. “Sentinel” circulating tumor cells allow early diagnosis of lung cancer in patients with chronic obstructive pulmonary disease. PloS one. 2014;9:e111597
85. Cilli A, Ozkaynak C, Onur R. et al. Lung cancer detection with low-dose spiral computed tomography in chronic obstructive pulmonary disease patients. Acta Radiol. 2007;48:405-11
86. Vaupel P, Mayer A. Hypoxia in tumors: pathogenesis-related classification, characterization of hypoxia subtypes, and associated biological and clinical implications. Adv Exp Med Biol. 2014;812:19-24
87. Lambert AW, Pattabiraman DR, Weinberg RA. Emerging Biological Principles of Metastasis. Cell. 2017;168:670-91
88. Hamilton G, Rath B. Circulating tumor cell interactions with macrophages: implications for biology and treatment. Transl Lung Cancer Res. 2017;6:418-30
89. Pleasance ED, Stephens PJ, O'Meara S. et al. A small-cell lung cancer genome with complex signatures of tobacco exposure. Nature. 2010;463:184-90
90. Fischer KR, Durrans A, Lee S. et al. Epithelial-to-mesenchymal transition is not required for lung metastasis but contributes to chemoresistance. Nature. 2015;527:472-6
91. Liu S, Zhou F, Shen Y. et al. Fluid shear stress induces epithelial-mesenchymal transition (EMT) in Hep-2 cells. Oncotarget. 2016;7:32876-92
92. Stoecklein NH, Klein CA. Genetic disparity between primary tumours, disseminated tumour cells, and manifest metastasis. Int J. Cancer. 2010;126:589-98
93. Minder P, Zajac E, Quigley JP, Deryugina EI. EGFR regulates the development and microarchitecture of intratumoral angiogenic vasculature capable of sustaining cancer cell intravasation. Neoplasia. 2015;17:634-49
94. Naumov GN, MacDonald IC, Chambers AF, Groom AC. Solitary cancer cells as a possible source of tumour dormancy? Semin Cancer Biol. 2001;11:271-6
95. Wong CW, Song C, Grimes MM. et al. Intravascular location of breast cancer cells after spontaneous metastasis to the lung. Am J Pathol. 2002;161:749-53
96. Hamilton G, Rath B, Holzer S. et al. Second-line therapy for small cell lung cancer: exploring the potential role of circulating tumor cell. Transl Lung Cancer Res. 2016;5:71-7
97. Naxerova K, Reiter JG, Brachtel E. et al. Origins of lymphatic and distant metastases in human colorectal cancer. Science. 2017;357:55-60
98. Smerage JB, Hayes DF. The measurement and therapeutic implications of circulating tumour cells in breast cancer. Br J Cancer. 2006;94:8-12
99. Semenova EA, Nagel R, Berns A. Origins, genetic landscape, and emerging therapies of small cell lung cancer. Genes Dev. 2015;29:1447-62
100. Hamilton G, Rath B, Klameth L. et al. Small cell lung cancer: Recruitment of macrophages by circulating tumor cells. Oncoimmunology. 2016;5:e1093277
101. Fiori ME, Villanova L, De Maria R. Cancer stem cells: at the forefront of personalized medicine and immunotherapy. Curr Opin Pharmacol. 2017;35:1-11
102. Hamilton G, Rath B. Mesenchymal-Epithelial Transition and Circulating Tumor Cells in Small Cell Lung Cancer. Adv Exp Med Biol. 2017;994:229-45
103. Steeg PS. Targeting metastasis. Nature Rev Cancer. 2016;16:201-18
104. Meikle CK, Kelly CA, Garg P. et al. Cancer and Thrombosis: The Platelet Perspective. Front Cell Dev Biol. 2017;4:147
105. Eifler RL, Lind J, Falkenhagen D. et al. Enrichment of circulating tumor cells from a large blood volume using leukapheresis and elutriation: proof of concept. Cytometry B Clin Cytom. 2011;80:100-11
106. Fischer JC, Niederacher D, Topp SA. et al. Diagnostic leukapheresis enables reliable detection of circulating tumor cells of nonmetastatic cancer patients. Proc Natl Acad Sci U S A. 2013;110:16580-5
107. Hosseini H, Obradović MMS, Hoffmann M. et al. Early dissemination seeds metastasis in breast cancer. Nature. 2016;540:552-8
108. Wu T, Cheng B, Fu L. Clinical Applications of Circulating Tumor Cells in Pharmacotherapy: Challenges and Perspectives. Mol Pharmacol. 2017;92:232-9
Author contact
![]() Corresponding author: Gerhard Hamilton. Ph.D., Office +431-40400-41700, hamilton.srbtcorg or gerhard.hamiltonac.at
Corresponding author: Gerhard Hamilton. Ph.D., Office +431-40400-41700, hamilton.srbtcorg or gerhard.hamiltonac.at