Oncomedicine 2018; 3:67-73. doi:10.7150/oncm.25500 This volume Cite
Research Paper
BP1 Regulates PI3K/Akt Pathway by Targeting Vascular Endothelial Growth Factor (VEGF) in Breast Cancer
Jenny Hottel1, Caiqiao Yin1,2, Yi Lian1, Robert Siegel3, Xiaohui Tan1, Sidney W. Fu1 ![]()
1. Department of Medicine (Division of Genomic Medicine), and Department of Microbiology, Immunology and Tropical Medicine, The George Washington University School of Medicine and Health Sciences, Washington, DC, USA
2. Affiliated Minda Hospital of Hubei University of Nationalities, Enshi, Hubei 445000, China
3. Department of Medicine (Division of Hematology/Oncology), The George Washington University School of Medicine and Health Sciences, Washington, DC, USA
Received 2018-2-11; Accepted 2018-7-29; Published 2018-8-30
Abstract

The progression of breast cancer has been linked to a splice variant of DLX4 homeobox transcription factor called BP1, which is overexpressed in 81% of primary breast cancer. Our ChIP-on-chip and bioinformatic analysis studies showed that VEGFA is a potential target of BP1, and it is upregulated in cells overexpressing BP1. VEGFA has been implicated in tumor generation because it stimulates angiogenesis, proliferation, and migration. This study aims to decipher the functional roles of BP1 in VEGFA-PI3K/AKT pathways in tumorigenesis, using an estrogen receptor (ER) negative breast cancer cell line, Hs578T. QPCR and Western blots were performed on three overexpressing and two vector control cell lines to assess and compare the transcriptional and translational expression. Our findings illustrate the increased expression of VEGFA, pPI3K, and pAkt when BP1 is overexpressed. Therefore, BP1 may play an important role in regulating the PI3K/Akt pathway through VEGFA.
Keywords: breast cancer, BP1, VEGFA and PI3K
Introduction
Homeobox gene is composed of a highly conserved 180 base pairs of DNA fragment and involved in cell differentiation, growth, and embryonic development, encompassing many species. Homeobox genes encode transcription factors that regulate downstream targets. Those encoded homeodomains bind DNA through a helix-turn-helix structure and are about sixty amino acids in length. Typically these homeodomain proteins along with other transcription factors activate or repress genes by simultaneously targeting the promoter region in complexes. This is due to the lack of DNA binding specificity of a single homeoprotein, which becomes much more effective when combined with additional transcription factors.
At least 300 human homeobox genes have been described, resulting in 253 functional genes and 65 pseudogenes(1). They are classified into 11 homeobox gene classes and further subdivided into 102 homeobox gene families(1). The largest homeobox gene family is the HOX gene family and the human genome contains 39 HOX genes divided into four clusters(1). Other families include Dlx, Evx, Msx, Nkx, Pax, Cdx, En, Otx, Pitx, and Emx and many others(1).
Since HOX genes play an important role in embryogenesis, alterations in the expression of those genes can cause major developmental disruptions. The deregulated expression of homeobox genes has been described in many solid tumors and derivative cell lines. Homeobox genes are expressed in carcinoma cell lines, as well as the corresponding embryonic tissues from which these tumor cells are derived (2) (3). Deregulation of homeobox genes in breast cancer clearly indicates a role for these genes in cellular transformation, alterations in cell cycle and apoptosis, and progression to a metastasis. Some of the classes involved in promoting cellular transformation in breast cancer include HOX, HSIX1, and PAX(2) (4) (5). A HOX gene detected in breast cancer lesions and surrounding tissues is HOXA1(6). While HOX genes like HOXA5 (7) and HOXA9 (10) have been implicated in breast cancer because of loss of function. Also within the distal-less (DLX) family of homeobox genes, including DLX4, there is evidence of overexpression in 81% of primary breast cancers (8). There are at least seven members in the DLX family and DLX4 has at least two isoforms, including variant 1, also called BP1, and variant 2, sometimes refer to as DLX7, and each has its own distinct functions (9).
Cascade between VEGFA, a direct target of BP1, and its downstream target genes in cell proliferation by Pathway Studio 5.0. Gene network showing the direct binding of BP1 to VEGF, activating cascades to promote cell proliferation. Upregulated genes are in red, while downregulated genes are in green. (15)
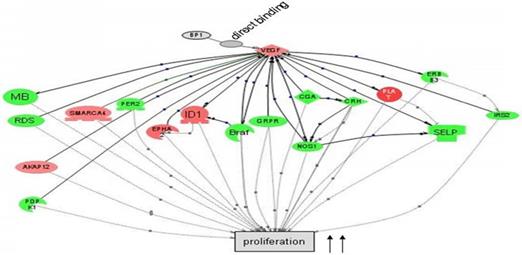
BP1, as an isoform of DLX4, functions as a transcription factor that binds to two silencer sequences upstream of the β-globulin gene (9) (10). In cancer cell lines BP1 mRNA and protein are expressed at increased levels (11). Also its expression has been correlated with the progression of breast cancer (12), possibly providing those cancer cells with a survival advantage (11). Current studies also showed that overexpression of BP1 presents a more aggressive phenotype in both MCF7 and Hs 578T cells (12). Furthermore, there is correlation between increased BP1 expression and tumorigenic potential in breast cancer cell lines (8). BP1 is also linked to increased expression of integrins, which have also been implicated in contributing to inducing angiogenesis (13, 14).
In preceding experiments to discover possible markers in pathways relevant to BP1 expression, a BP1 ORF-carrying plasmid was transfected into Hs578T cells and three stable cell lines were established along with two empty vector-containing cell lines as controls. After the microarray study and data mining, we have identified a number of a number of relevant pathways that are associated with BP1 overexpression. Figure 1 depicts that BP1 binds to its target, VEGF and impacts cell proliferation.
DLX4 has also been shown to induce expression of vascular endothelial growth factor (VEGF), which is a heparin binding homodimeric glycoprotein, in ovarian carcinomas (16). VEGF acts through endothelial specific receptor tyrosine kinases, VEGFR1, VEGFR2, and VEFGR3. Among those, VEGFR2 is the major signal transducing receptor for angiogenesis of endothelial cells. Also VEGFA is shown to be a direct target of BP1 and is upregulated by BP1 expression (15). These findings indicate that further analysis of BP1 regulation of VEGFA is required in the search for a therapeutic target that possesses both clinical usefulness in terms of prognostic prediction in cancer and clear functional relevance for studying cancer biology in relation to tumorigenesis and metastasis.
VEGFA is integral to the formation of vasculature in response to wounds and tumors. As endothelial cells are activated to form new blood vessels, by the binding of angiogenic growth factors to their receptors, they require the aide of integrins in the extension of the newly formed blood vessels (17). VEGFA has been shown to be a direct ligand of integrin α9β1 and mediate cell interactions with integrins α3β1 and αvβ3 (13, 18). VEGFA can induce angiogenesis in tumor cells as well as surrounding cells, generating an increased proliferative ability. The unbalanced secretion of these cytokines results in the creation of blood vessels that are disorganized and abnormal in their structure and function (19, 20). Tumor suppressors and oncogenes mediate tumor growth by their ability to induce overexpression of VEGFA (21, 22). In the majority of carcinomas, tumors, sarcomas, and even some pre-malignant lesions VEGFA is overexpressed (19, 23-25). These expression levels increase as the cancer progresses (17).
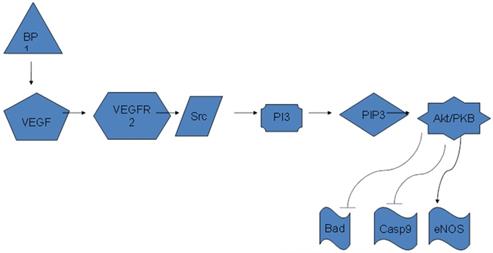
VEGFA is regulated in many ways, including the MAPK pathway, hypoxia, hormones, and oncogenes. Hypoxic areas develop during tumor growth leading to the increased expression of VEGF. This expression is mediated through HIF-1α, which is driven by the hypoxic conditions, resulting in an increase in VEGF transcription (17). Hormones, like estrogens and progestins, activate VEGF transcription by binding to estrogen response elements (26, 27). Also its transcription can be induced by prolactin in a MAPK and Jak2 dependent manner (27, 28). Increased expression of VEGF has been noted from oncogenes such as Ras, Src, HER2/neu, and Bcr/Abl(27, 29, 30). The downstream progression of the VEGF pathway is shown in Figure 1, with emphasis on signaling and interactions promoting angiogenesis. The cascade to the far left is of particular interest, as cell survival is mediated through PI3K (phosphatidylinositol 3-kinasae) and Akt/PKB (Protein Kinase-B) among others. Akt/PKB is a kinase that engages in many cell signaling avenues, including those promoting cell growth, inhibition of apoptosis, metabolism, and angiogenesis. ADAM17, a shedding enzyme, has been shown to promote breast cancer through the stimulation of PI3K and Akt (31). While in an ER positive cell line, MCF-7, studies have shown keratinocyte growth factor (KGF) to upregulate mRNA expression and increase Akt phosphorylation (32). Conversely the expression was blocked by inhibiting the PI3k/Akt pathway with LY, a PI3 kinase inhibitor (32).
In view of the fact that VEGFA plays a critical role in tumor biology, it along with its receptors has been identified as targets for cancer therapy. At present there are several drugs available to block VEGFA or inhibit its receptors. Bevacizumab is an anti-VEGF drug, while sunitinib and sorafenib are anti-VEGF receptor (17). Many tumors do not respond well to anti-VEGF treatment because there are other angiogenic pathways (33). Thus looking for upstream targets is warranted.
Since VEGFA has shown to be upregulated in breast cancer cells by BP1 and in ovarian cancer cells by DLX4, there is vast potential for reducing angiogenesis by targeting players upstream of VEGFA (15, 16). The purpose of this study is to better define the tumorigenic relationship of BP1 with its target VEGFA and thusly a connection to the PIP3/Akt pathway. Further investigation of this pathway at the transcriptional and translational level is essential to discovering how to possibly decrease angiogenesis, proliferation, and tumor metastasis through suppression of BP1 to obstruct the connection between them. Characterization of this interaction could lead to unearthing new targets for breast cancer intervention and early detection.
Materials and Methods
Cell culture
BP1-transfected Hs578T breast cancer stable lines, previously established in our lab, were cultured in DMEM medium (Invitrogen Corporation, Carlsbad, CA, USA) with 10% fetal bovine serum (Atlanta Biologicals, Lawrenceville, GA, USA), penicillin 50 unit/ml-streptomycin 50μg/ml (Invitrogen Corporation, Carlsbad, CA, USA), and G418 sulfate added (Invitrogen Corporation, Carlsbad, CA, USA). They were incubated at 37ºC humidified incubator supplemented with 5% CO2.
RNA Isolation, cDNA Synthesis and Quantitative Real-time PCR
Total RNA was extracted from Hs 578T cell pellets using Trizol reagent (Invitrogen Corporation, Carlsbad, CA, USA) and the concentration determined by an ultra violet spectrophotometer at 260nm (Model DU 640B, Beckman, Fullerton, CA, USA). There are total of five samples, three BP1 overexpressing cell lines (O3, O6 and O7) and two vector control cell lines (V1 and V2). The iScript cDNA synthesis kit (Bio-Rad Laboratories, Hercules, CA, USA) was used and 1ug of total RNA for each sample, 4μl of 5x reaction master mix, 1μl of iScript reverse transcriptase brought to a total volume of 20μl with nuclease free water. The incubation conditions using a thermal cycler (Model 2720, Applied Biosystems, Foster City, CA, USA) were as follows: 5 min. at 25ºC, 30 min. at 42ºC, 5 min. at 85ºC, and hold at 4ºC. The synthesized cDNA was diluted (1:5) before amplifying the gene targets using the Power SYBR Green PCR Master Mix kit (Applied Biosystems, Foster City, CA, USA). Diluted cDNA, 1μl, was added to 12.5μl of master mix, 1μl of each forward and reverse primers (Integrated DNA Technologies, Coralville, IA, USA) for a total volume of 25μl in nuclease free water. Samples were run in triplicate on a 7300 Real-Time PCR system (Applied Biosystems, Foster City, CA, USA) with the following parameters: stage one 2 min. at 50ºC, 10 min. 95ºC; stage two 15 sec. at 95ºC, 1 min. at 60ºC; stage three 15 sec. at 95ºC, 1 min. at 60ºC, 15 sec. at 95ºC, 15sec. at 60ºC. Primers were designed using NCBI/Primer-BLAST and the sequences are shown in Table 1. Expression was normalized against ribosomal 18S and the average Ct values were used to calculate expression levels based on the log2 transformation values.
Protein Isolation
Cells were harvested by adding 0.25% trypsin-EDTA (Mediatech Incorporated, Manassas, VA, USA) and incubated for 2 minutes at 37ºC when reached 75-95% confluence. The cells were then washed with 1x PBS before being pelleted for 5 minutes at 3,000 rpm. Pellets were resuspended in 300μl of a protease inhibitor / tissue protein extraction agent (T-PER) combination (Pierce, Rockford, IL, USA). After centrifuging for another 5 minutes at 10,000 rpm the protein rich supernatant was stored at -80ºC.
Primer Sequences used for QPCR.
| Target Gene | Sequence |
|---|---|
| VEGFA | Forward 5'-ctacctccaccatgccaagt-3' |
| Reverse 5'-gcagtagctgcgctgataga-3' | |
| VEGFR2 | Forward 5'-GACTTGGCCTCGGTCATTTA-3' |
| Reverse 5'-ACACGACTCCATGTTGGTCA-3' | |
| Src | Forward 5'-ACTGAGCTCACCACAAAGGG-3' |
| Reverse 5'-CACTGGCACATGAGGTCGT-3' | |
| PI3K | Forward 5'-GAAAAACGAGGAATCCGACA-3' |
| Reverse 5'-TCAAAGGCAGGGTTACTCCA-3' | |
| Akt1 | Forward 5'-AGAAGCAGGAGGAGGAGGAG-3' |
| Reverse 5'-CCCAGCAGCTTCAGGTACTC-3' | |
| Akt2 | Forward 5'-CCGAGGTCGACACAAGGTA-3' |
| Reverse 5'-CTGGTCCAGCTCCAGTAAGC-3' | |
| Akt3 | Forward 5'-CGTGGCAAAATGAACACATC-3' |
| Reverse 5'-TGATCCCAAGAAACACACCA-3' |
Western Blot Assays
For gel electrophoresis, a total of 30ug of the protein sample was loaded for each sample. The samples were brought to 20μl with T-PER and combined with 4μl of 6x SDS dye, for a total of 24μl per well. They were then incubated at 98ºC for 5 min. before loading onto a 4-15% Tris-HCl ready gel (Bio-Rad Laboratories, Hercules, CA, USA) along with 5μl of PageRuler Plus Prestained Protein Ladder (Fermentas Incorporated, Glen Burnie, MD, USA). The gel was run at 90V for two hours in 1x running buffer, which contains Tris-HCL/Glycine/SDS in a liter of deionized water (Fisher Scientific, Fair Lawn, NJ, USA). Upon completion the gel was removed and incubated in the transfer buffer. Polyscreen PVDF transfer membrane (PerkinElmer Life Sciences, Boston, MA, USA) was soaked in methanol (Fisher Scientific, Fair Lawn, NJ, USA), while the sponges and filter paper (Sigma-Aldrich, St. Louis, MO, USA) were soaked in 1x Tris/Glycine transfer buffer (Bio-Rad Laboratories, Hercules, CA, USA). The transfer sandwich was then set up. The transfer was completed overnight at 4ºC with 80 mAmp of current. After transfer, the membrane was washed twice for 15 min. each in 1x PBS with 1% Tween 20 (Fisher Scientific, Fair Lawn, NJ, USA) with shaking. The membrane was blocked in 5% non-fat dry milk (Nestle Carnation, Vevey, Switzerland) in 1x PBST for two hrs. at room temperature with shaking. Again the membrane is washed twice for 15 min. in 1x PBST. Primary antibody in 1x PBST is then added at the appropriate dilution and incubated overnight at 4ºC with shaking. The blots were probed with four different antibodies, including actin. The two phosphorylated antibodies were incubated with 5% non-fat dry milk in 1x PBST. The membrane is washed twice for 15 min. each in 1x PBST and then the secondary antibody is added using the appropriate dilution in 1x PBST for two hrs. at room temperature with shaking. After another set of washes the next step is visualization with SuperSignal West Pico Maximum Sensitivity Substrate (Thermo Scientific, Rockford, IL, USA). The A and B solutions are mixed in equal parts, 700μl each, and then applied to the membrane, which is then incubated in the dark for five minutes before exposure with a Kodak 2000MM Image System (Kodak, Rochester, NY, USA). Lastly the membrane was stripped at 60ºC with shaking using a solution of Tris-HCL/SDS/β-mercaptoethanol. The blot was then reused starting over from the first set of washes.
Statistical analysis
Gene expression was analyzed by the student t-test. P-values less than 0.05 were considered statistically significant.
Results
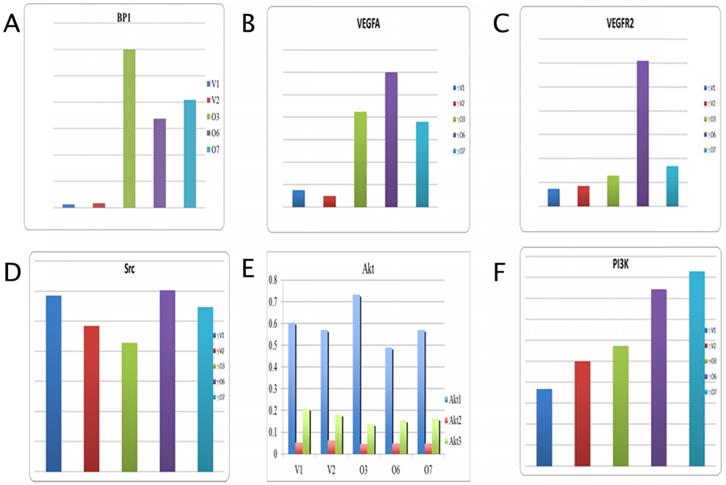
To determine the level of gene expression in the Hs 578T cells at the transcriptional level, qPCR was performed for five different genes in the cascade for the PI3k/Ak pathway, including VEGFA, VEGFR2, Src, PI3K, and 3 transcript variants of Akt. All data was normalized against 18S and analyzed to determine differences in fold changes.
In Figure 2A, the expression level for BP1 is provided for reference and shows a 15 fold increase in overexpressing cell lines by average. Data analysis for VEGFA shows a marked increase in expression in the BP1 overexpressing cell lines compared to the vector controls, an average change of 5.74 (Figure 2B). While vascular endothelial growth factor receptor two (VEGFR2) gives a difference in fold change of 4.14 between overexpressors and controls (Figure 2C). There is no significant change in Src expression, with a fold change value of 0.987 (Figure 2D). All three transcript variants of the Akt gene show little difference in their mRNA expression between the BP1 overexpressing cell lines and the vector controls. However the expression levels for Akt1 are more even keeled, save for O3, than that of Akt2 and Akt3.
Those two transcripts actually show the control cell lines with higher expression than that of the BP1 overexpressing lines (Figure 2E). For Akt1, there is a difference in fold change of 1.01, while for Akt2 the difference is .754. Lastly Akt3 shows a fold change of .80. Thus there's no statistical significance for the expression of Src and the Akt transcripts. The overexpressors show greater expression compared to the vector controls for the PI3K gene, with a fold change of 1.93 (Figure 2F).
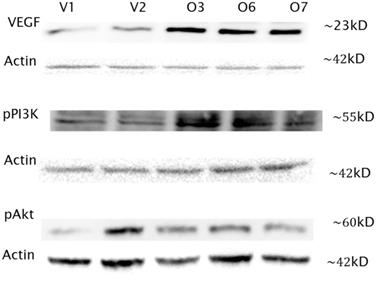
In addition to the transcripts analysis, Western blot assays were performed for three of the genes in the PI3K/Akt pathway to assess the protein expression. All protein expression levels were normalized with actin (Santa Cruz Biotechnology Incorporated, Santa Cruz, CA, USA). VEGF expression is clearly elevated by average fold change of 5.9, as shown in Figure 3, with the expected band size of 23kD for VEGF. Protein levels in the overexpressors were higher than those of the vector controls. Next phosphorylated PI3K was probed for and bands were noted at 55kD as expected (Figure 3). However no bands were seen at 85kD, which the antibody used could also detect. The overexpressing cell lines showed increased PI3K expression by 1.41 fold. Finally phosphorylated Akt was detected at 60kD. While the overexpressors showed an increase over V1 one but not V2, differing by a fold change of 1.59. The protein expression levels are in consistent to their respective transcript levels, although they are not necessarily always in agreement.
qPCR Analysis for BP1, VEGFA, VEGFR, Src, PI3K, Akt1, Akt2, and Akt3 expression (y-axis) in Hs 578T transfected cell lines (V1, V2 vs. O3, O6 and O7). The expression data was normalized with 18S. The fold change is about 15 for BP1, 6 for VEGFA, 4 for VEGFR2 and 2 for PI3K. For Akt1 the vector controls and overexpressors are fairly equal, with a difference of 1.01. There were no significant change between overexpressors and controls in Akt1, Akt2 and Akt3 expression.
Discussion
It has been shown that BP1, a splice variant of DLX4 transcription factor, is overexpressed in breast cancer cells and that its expression is associated with breast cancer progression(12). VEGFA has been implicated in tumor generation because it stimulates angiogenesis, proliferation, and migration(20). Also of note is that BP1 directly binds to VEGFA, which is subsequently upregulated(15) (Figure 4). VEGF, along with its receptors have been linked to angiogenesis through the PI3K/Akt pathway(17) (34, 35). Whereas angiogenesis is considered to be an important target for breast cancer therapy(36). These findings strongly suggest that BP1 may play an important role upstream of the PI3K/Akt pathway, which has a significant role in cell survival and angiogenesis, thus presenting BP1 as a possible candidate as a therapeutic target in breast cancer.
Western Blot analysis of VEGF, pPI3K and pAkt in Hs 587T BP1 overexpressors compared to vector controls. Three overexpressing cell lines (O3, O6 and O7) and two vector control cells lines (V1, V2) were analyzed by Western blot with VEGF, pPI3K and pAkt antibody respectively. Actin was used to normalize protein expression levels. VEGF and pPI3K expression was increased in the overexpressing cells by a difference in fold change of 5.9 and 1.41 respectively compared to the vector controls. pAkt expression was increased in the overexpressing cells compared to O1 by 1.59, but O2.
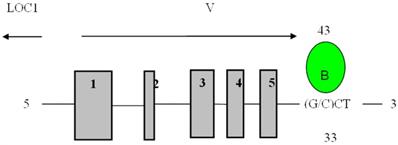
Direct binding site of BP1 to VEGFA obtained from the UCSC Genome Browser. Transcript variant six of VEGFA is shown with BP1 binding downstream of the coding region, 3313bp away from the transcription start site. The binding site is in the untranslated region (UTR) at 43849244. (15)
Three BP1-overexpressing cell lines and two vector controls of the Hs578T cell line were used for the current study. Quantitative RT-PCR showed differing accounts of the level of mRNA expression among the genes assayed. The data for VEGFA showed the greatest potential with results confirming that the expression levels of the overexpressors are increased, with a difference in fold change of 5.74, while VEGFR2 showed a 4.14 fold difference. However, we noticed that O6 cell line showed an exceptionally high VEGFR2 expression, which is worth further analysis. The other five transcripts, Src, pPI3K, Akt1, Akt2, Akt3, show virtually no change between the overexpressors and the controls, which indicates that BP1 does not directly regulate their expression at least at the transcriptional level.
Diagram of BP1 regulating Akt/PI3K pathway through VEGFA. BP1 binds to the 3'-UTR of VEGFA and upregulates its expression. VEGFA in turn activates the PI3K/Akt pathway to promote cell proliferation.
The major players in the PI3K/Akt pathway were chosen to be scrutinized for their protein expression, alongside of the key connector VEGF. The Western analysis shows VEGF protein levels in the overexpressing cell lines to be notably higher than those of the vector controls, a difference in fold change of 5.9, which is consistent with our qPCR data. For phosphorylated PI3K, the overexpressors show higher expression than the controls. The protein expression levels between the overexpressors and the vectors differ by a fold change of 1.41 although we did not see the same level of changes in mRNA. The expression for phosphorylated Akt was increased in all three overexpressors as compared to V1. However V2 had the highest yield of all, unexpectedly. This could be an anomaly to what was predicted or due to a concentration discrepancy or a gel loading error. The difference in fold change between the overexpressors and V1 is 1.59. This could be due to the complexity or effect of other components or mechanisms along the pathway not evaluated. In addition, the correlation between mRNA and protein abundances in the cell has been reported to be notoriously poor due to the complex biology.
These results showcase how BP1 upregulates VEGF, which in turn upregulates key members of the PI3K/Akt pathway (Figure 5). Additional members not surveyed in this study, such as Bad and Caspase9, should be investigated in future studies to give a complete analysis of the relationship between BP1, VEGF, and its downstream targets, including the end products. The activation of this pathway contributes to cell survival and angiogenesis. These are both factors in the progression and development of breast cancer. Since BP1 is an upstream transcriptional regulator, it could be considered as a new target in the treatment and prevention of breast cancer.
Conclusions
This study demonstrates the relationship between BP1 and its downstream target VEGF, and VEGF-related PI3K/Akt pathway. Analysis of key elements at the transcriptional and translational level illustrates the increased expression of VEGFA, pPI3K, and pAkt when BP1 is overexpressed. Our findings implicate BP1 plays an important role in the PI3K/Akt pathway in increased cell survival and angiogenesis. Further analysis of this pathway and its components would solidify BP1, possibly through its suppression, as a contender for therapy in breast cancer treatment.
Acknowledgements
This research was supported by the Elaine H. Snyder Cancer Research Award (to SWF).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Holland PW, Booth HA, Bruford EA. Classification and nomenclature of all human homeobox genes. BMC Biol. 2007;5:47
2. Peverali FA, D'Esposito M, Acampora D, Bunone G, Negri M, Faiella A. et al. Expression of HOX homeogenes in human neuroblastoma cell culture lines. Differentiation. 1990;45:61-9
3. Cillo C, Barba P, Freschi G, Bucciarelli G, Magli MC, Boncinelli E. HOX gene expression in normal and neoplastic human kidney. Int J Cancer. 1992;51:892-7
4. Silberstein GB, Van Horn K, Hrabeta-Robinson E, Compton J. Estrogen-triggered delays in mammary gland gene expression during the estrous cycle: evidence for a novel timing system. J Endocrinol. 2006;190:225-39
5. Ford HL, Kabingu EN, Bump EA, Mutter GL, Pardee AB. Abrogation of the G2 cell cycle checkpoint associated with overexpression of HSIX1: a possible mechanism of breast carcinogenesis. Proc Natl Acad Sci U S A. 1998;95:12608-13
6. Chariot A, Castronovo V. Detection of HOXA1 expression in human breast cancer. Biochem Biophys Res Commun. 1996;222:292-7
7. Raman V, Martensen SA, Reisman D, Evron E, Odenwald WF, Jaffee E. et al. Compromised HOXA5 function can limit p53 expression in human breast tumours. Nature. 2000;405:974-8
8. Fu SW, Schwartz A, Stevenson H, Pinzone JJ, Davenport GJ, Orenstein JM. et al. Correlation of expression of BP1, a homeobox gene, with estrogen receptor status in breast cancer. Breast Cancer Res. 2003;5:R82-7
9. Fu S, Stevenson H, Strovel JW, Haga SB, Stamberg J, Do K. et al. Distinct functions of two isoforms of a homeobox gene, BP1 and DLX7, in the regulation of the beta-globin gene. Gene. 2001;278:131-9
10. Chase MB, Fu S, Haga SB, Davenport G, Stevenson H, Do K. et al. BP1, a homeodomain-containing isoform of DLX4, represses the beta-globin gene. Mol Cell Biol. 2002;22:2505-14
11. Stevenson HS, Fu SW, Pinzone JJ, Rheey J, Simmens SJ, Berg PE. BP1 transcriptionally activates bcl-2 and inhibits TNFalpha-induced cell death in MCF7 breast cancer cells. Breast Cancer Res. 2007;9:R60
12. Man YG, Fu SW, Schwartz A, Pinzone JJ, Simmens SJ, Berg PE. Expression of BP1, a novel homeobox gene, correlates with breast cancer progression and invasion. Breast Cancer Res Treat. 2005;90:241-7
13. Vlahakis NE, Young BA, Atakilit A, Hawkridge AE, Issaka RB, Boudreau N. et al. Integrin alpha9beta1 directly binds to vascular endothelial growth factor (VEGF)-A and contributes to VEGF-A-induced angiogenesis. J Biol Chem. 2007;282:15187-96
14. Boudreau NJ, Varner JA. The homeobox transcription factor Hox D3 promotes integrin alpha5beta1 expression and function during angiogenesis. J Biol Chem. 2004;279:4862-8
15. Song Y, Dang C, Fu Y, Lian Y, Hottel J, Li X. et al. Genome-wide analysis of BP1 transcriptional targets in breast cancer cell line Hs578T. Int J Biol Sci. 2009;5:1-12
16. Hara F, Samuel S, Liu J, Rosen D, Langley RR, Naora H. A homeobox gene related to Drosophila distal-less promotes ovarian tumorigenicity by inducing expression of vascular endothelial growth factor and fibroblast growth factor-2. Am J Pathol. 2007;170:1594-606
17. Balan B, Slotwinski, R. VEGF and tumor angiogenesis. Central European Journal of Immunology. 2008;33:232-6
18. Hutchings H, Ortega N, Plouet J. Extracellular matrix-bound vascular endothelial growth factor promotes endothelial cell adhesion, migration, and survival through integrin ligation. FASEB J. 2003;17:1520-2
19. Dvorak HF. Vascular permeability factor/vascular endothelial growth factor: a critical cytokine in tumor angiogenesis and a potential target for diagnosis and therapy. J Clin Oncol. 2002;20:4368-80
20. Nagy JA, Dvorak AM, Dvorak HF. VEGF-A and the induction of pathological angiogenesis. Annu Rev Pathol. 2007;2:251-75
21. Rak J, Yu JL. Oncogenes and tumor angiogenesis: the question of vascular "supply" and vascular "demand". Semin Cancer Biol. 2004;14:93-104
22. Mukhopadhyay D, Datta K. Multiple regulatory pathways of vascular permeability factor/vascular endothelial growth factor (VPF/VEGF) expression in tumors. Semin Cancer Biol. 2004;14:123-30
23. Brown LF, Detmar M, Claffey K, Nagy JA, Feng D, Dvorak AM. et al. Vascular permeability factor/vascular endothelial growth factor: a multifunctional angiogenic cytokine. EXS. 1997;79:233-69
24. Mentzel T, Brown LF, Dvorak HF, Kuhnen C, Stiller KJ, Katenkamp D. et al. The association between tumour progression and vascularity in myxofibrosarcoma and myxoid/round cell liposarcoma. Virchows Arch. 2001;438:13-22
25. Guidi AJ, Schnitt SJ, Fischer L, Tognazzi K, Harris JR, Dvorak HF. et al. Vascular permeability factor (vascular endothelial growth factor) expression and angiogenesis in patients with ductal carcinoma in situ of the breast. Cancer. 1997;80:1945-53
26. Hyder SM, Huang JC, Nawaz Z, Boettger-Tong H, Makela S, Chiappetta C. et al. Regulation of vascular endothelial growth factor expression by estrogens and progestins. Environ Health Perspect. 2000;108(Suppl 5):785-90
27. Birk DM, Barbato J, Mureebe L, Chaer RA. Current insights on the biology and clinical aspects of VEGF regulation. Vasc Endovascular Surg. 2008;42:517-30
28. Goldhar AS, Vonderhaar BK, Trott JF, Hovey RC. Prolactin-induced expression of vascular endothelial growth factor via Egr-1. Mol Cell Endocrinol. 2005;232:9-19
29. Rak J, Filmus J, Finkenzeller G, Grugel S, Marme D, Kerbel RS. Oncogenes as inducers of tumor angiogenesis. Cancer Metastasis Rev. 1995;14:263-77
30. Mukhopadhyay D, Tsiokas L, Zhou XM, Foster D, Brugge JS, Sukhatme VP. Hypoxic induction of human vascular endothelial growth factor expression through c-Src activation. Nature. 1995;375:577-81
31. Zheng X, Jiang F, Katakowski M, Zhang ZG, Lu QE, Chopp M. ADAM17 promotes breast cancer cell malignant phenotype through EGFR-PI3K-AKT activation. Cancer Biol Ther. 2009;8:1045-54
32. Chang HL, Sugimoto Y, Liu S, Wang LS, Huang YW, Ye W. et al. Keratinocyte growth factor (KGF) regulates estrogen receptor-alpha (ER-alpha) expression and cell apoptosis via phosphatidylinositol 3-kinase (PI3K)/Akt pathway in human breast cancer cells. Anticancer Res. 2009;29:3195-205
33. Yoshiji H, Harris SR, Thorgeirsson UP. Vascular endothelial growth factor is essential for initial but not continued in vivo growth of human breast carcinoma cells. Cancer Res. 1997;57:3924-8
34. Cebe-Suarez S, Zehnder-Fjallman A, Ballmer-Hofer K. The role of VEGF receptors in angiogenesis; complex partnerships. Cell Mol Life Sci. 2006;63:601-15
35. Liang Z, Brooks J, Willard M, Liang K, Yoon Y, Kang S. et al. CXCR4/CXCL12 axis promotes VEGF-mediated tumor angiogenesis through Akt signaling pathway. Biochem Biophys Res Commun. 2007;359:716-22
36. Hayes DF, Miller K, Sledge G. Angiogenesis as targeted breast cancer therapy. Breast. 2007;16(Suppl 2):S17-9
Author contact
![]() Corresponding author: Sidney W. Fu, M.D., Ph.D., Department of Medicine, Division of Genomic Medicine, The George Washington University School of Medicine and Health Sciences, 2300 Eye Street, N.W. Ross Hall 402C Washington, DC 20037. Email: sfuedu; Tel: 202-994-4767
Corresponding author: Sidney W. Fu, M.D., Ph.D., Department of Medicine, Division of Genomic Medicine, The George Washington University School of Medicine and Health Sciences, 2300 Eye Street, N.W. Ross Hall 402C Washington, DC 20037. Email: sfuedu; Tel: 202-994-4767